Pretransplant Comorbidities That Affect the Risk of Transplant-Related Mortality: Predictive Power of the Hematopoietic Cell Transplant–Specific Comorbidity Index
Because of the intensity of therapy associated with the transplant process, the pretransplant clinical status of recipients (e.g., age, presence of infections or organ dysfunction, and functional status) is associated with a risk of transplant-related mortality.
The best tool to assess the impact of pretransplant comorbidities on outcomes after transplant was developed by adapting an existing comorbidity scale, the Charlson Comorbidity Index (CCI). Investigators at the Fred Hutchinson Cancer Research Center systematically defined which of the CCI elements were correlated with transplant-related mortality in adult and pediatric patients. They also determined several additional comorbidities that have predictive power specific to transplant patients.
Successful validation defined what is now termed the hematopoietic cell transplant–specific comorbidity index (HCT-CI).[1,2] The rate of transplant-related mortality increases with the presence of cardiac, hepatic, pulmonary, gastrointestinal, infectious, and autoimmune comorbidities, or a history of previous solid tumors (see Table 1).
Table 1. Definitions of Comorbidities Included in the Hematopoietic Cell Transplant–Specific Comorbidity Index (HCT-CI)a| HCT-CI Score |
|---|
| 1 | 2 | 3 |
|---|
| AST/ALT = aspartate aminotransferase/alanine aminotransferase; DLCO = diffusion capacity of carbon monoxide; FEV1 = forced expiratory volume in one second; ULN = upper limit of normal. |
| a Adapted from Sorror et al.[1] |
| b One-or-more–vessel coronary artery stenosis requiring medical treatment, stent, or bypass graft. |
| Arrhythmia: Atrial fibrillation or flutter, sick sinus syndrome, or ventricular arrhythmias | Moderate pulmonary: DLCO and/or FEV1 66%–80% or dyspnea on slight activity | Heart valve disease: Excluding mitral valve prolapse |
| Cardiac: Coronary artery disease,b congestive heart failure, myocardial infarction, or ejection fraction ≤50% | Moderate/severe renal: Serum creatinine >2 mg/dL, on dialysis, or prior renal transplant | Moderate/severe hepatic: Liver cirrhosis, bilirubin >1.5 × ULN, or AST/ALT >2.5 × ULN |
| Cerebrovascular disease: Transient ischemic attack or cerebrovascular accident | Peptic ulcer: Requiring treatment | Prior solid tumor: Treated at any time in the patient's history, excluding nonmelanoma skin cancer |
| Diabetes: Requiring treatment with insulin or oral hypoglycemic agents but not diet alone | Rheumatologic: Systemic lupus erythematosus, rheumatoid arthritis, polymyositis, mixed connective tissue disease, or polymyalgia rheumatica | Severe pulmonary: DLCO and/or FEV1 <65% or dyspnea at rest or requiring oxygen |
| Hepatic, mild: Chronic hepatitis, bilirubin >ULN or AST/ALT >ULN to 2.5 × ULN | | |
| Infection: Requiring continuation of antimicrobial treatment after day 0 | | |
| Inflammatory bowel disease: Crohn disease or ulcerative colitis | | |
| Obesity: Body mass index >35 kg/m2 | | |
| Psychiatric disturbance: Depression or anxiety requiring psychiatric consult or treatment | | |
The predictive power of this index for both transplant-related mortality and overall survival (OS) is strong, with a hazard ratio of 3.54 (95% confidence interval [CI], 2.0–6.3) for nonrelapse mortality and 2.69 (95% CI, 1.8–4.1) for survival in patients with a score of 3 or higher, compared with those who have a score of 0. Although the original studies were performed with patients who received intense myeloablative approaches, the HCT-CI has also been shown to predict outcomes for patients receiving reduced-intensity and nonmyeloablative regimens.[3] It has also been combined with disease status [4] and Karnofsky score,[5] leading to even better prediction of survival outcomes. In addition, high HCT-CI scores (>3) have been associated with a higher risk of grades III to IV acute graft-versus-host disease.[6]
Most patients assessed in the HCT-CI studies have been adults, and the comorbidities listed are skewed toward adult diseases. Several studies have explored the relevance of this scale for pediatric and young adult recipients of hematopoietic stem cell transplant (HSCT).
Evidence (use of HCT-CI score in pediatrics):
- A retrospective cohort study was conducted at four large centers of pediatric patients (median age, 6 years) with a wide variety of both malignant and nonmalignant disorders.[7]
- The HCT-CI was predictive of both nonrelapse mortality and survival.
- The 1-year nonrelapse mortality rates were:
- 10% for patients with scores of 0.
- 14% for patients with scores of 1 to 2.
- 28% for patients with scores of 3 or higher.
- The 1-year OS rates were:
- 88% for patients with scores of 0.
- 67% for patients with scores of 1 to 2.
- 62% for patients with scores of 3 or higher.
- A second study included young adults (aged 16–39 years) and demonstrated the following:[8]
- Similar increases in mortality with higher HCT-CI scores.
- The nonrelapse mortality rates were 24% for patients with scores of 0 to 2 and 38% for patients with scores of 3 or higher.
- The OS rates were 46% for patients with scores of 0 to 2 and 28% for patients with scores of 3 or higher.
- A prospective validation of the HCT-CI through the Center for International Blood and Marrow Transplant Research included 23,876 patients, 1,755 of whom were children, who underwent transplant between 2007 and 2009. Patients' HCT-CI scores and outcomes were tracked.[9]
- Although adults treated with myeloablative regimens had increased mortality rates with HCT-CI scores of 1 or 2, pediatric patients did not have increased mortality rates until a score of 3 or higher was noted.
Most of the reported comorbidities in these studies were respiratory or hepatic conditions and infections.[7,8] In the adolescent and young adult study, patients with pre-HSCT pulmonary dysfunction were at particularly higher risk of poor outcomes, with a 2-year OS rate of 29%, compared with 61% in those with normal lung function before HSCT.[8]
References:
- Sorror ML, Maris MB, Storb R, et al.: Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 106 (8): 2912-9, 2005.
- ElSawy M, Storer BE, Pulsipher MA, et al.: Multi-centre validation of the prognostic value of the haematopoietic cell transplantation- specific comorbidity index among recipient of allogeneic haematopoietic cell transplantation. Br J Haematol 170 (4): 574-83, 2015.
- Sorror ML, Storer BE, Maloney DG, et al.: Outcomes after allogeneic hematopoietic cell transplantation with nonmyeloablative or myeloablative conditioning regimens for treatment of lymphoma and chronic lymphocytic leukemia. Blood 111 (1): 446-52, 2008.
- Sorror ML, Sandmaier BM, Storer BE, et al.: Comorbidity and disease status based risk stratification of outcomes among patients with acute myeloid leukemia or myelodysplasia receiving allogeneic hematopoietic cell transplantation. J Clin Oncol 25 (27): 4246-54, 2007.
- Sorror M, Storer B, Sandmaier BM, et al.: Hematopoietic cell transplantation-comorbidity index and Karnofsky performance status are independent predictors of morbidity and mortality after allogeneic nonmyeloablative hematopoietic cell transplantation. Cancer 112 (9): 1992-2001, 2008.
- Sorror ML, Martin PJ, Storb RF, et al.: Pretransplant comorbidities predict severity of acute graft-versus-host disease and subsequent mortality. Blood 124 (2): 287-95, 2014.
- Smith AR, Majhail NS, MacMillan ML, et al.: Hematopoietic cell transplantation comorbidity index predicts transplantation outcomes in pediatric patients. Blood 117 (9): 2728-34, 2011.
- Wood W, Deal A, Whitley J, et al.: Usefulness of the hematopoietic cell transplantation-specific comorbidity index (HCT-CI) in predicting outcomes for adolescents and young adults with hematologic malignancies undergoing allogeneic stem cell transplant. Pediatr Blood Cancer 57 (3): 499-505, 2011.
- Sorror ML, Logan BR, Zhu X, et al.: Prospective Validation of the Predictive Power of the Hematopoietic Cell Transplantation Comorbidity Index: A Center for International Blood and Marrow Transplant Research Study. Biol Blood Marrow Transplant 21 (8): 1479-87, 2015.
Hematopoietic Stem Cell Transplant (HSCT)–Related Acute Complications
Infectious Risks and Immune Recovery After Transplant
Defective immune reconstitution is a major barrier to successful HSCT, regardless of graft source.[1,2] Serious infections have accounted for a significant percentage (4%–20%) of late deaths after HSCT.[3]
Factors that can significantly slow immune recovery include the following:[4]
- Graft manipulation (removal of T cells).
- Stem cell source (slow recovery with cord blood).
- Chronic graft-versus-host disease (GVHD).
Figure 1 illustrates the immune defects, contributing transplant-related factors, and types and timing of infections that occur after allogeneic transplant.[5]
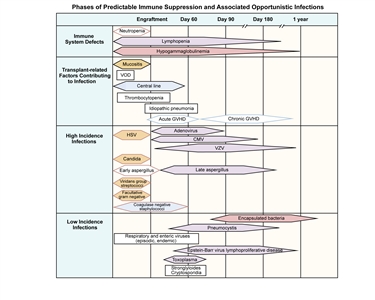
Figure 1. Phases of predictable immune suppression with their opportunistic infections among allogeneic hematopoietic stem cell transplant recipients. Adapted from Burik and Freifeld. This figure was published in Clinical Oncology, 3rd edition, Abeloff et al., Chapter: Infection in the severely immunocompromised patient, Pages 941–956, Copyright Elsevier (2004).
Bacterial infections tend to occur in the first few weeks after transplant during the neutropenic phase, when mucosal barriers are damaged from the conditioning regimen. There is significant ongoing research into the role of prophylactic antibacterial medications during the neutropenic phase.[6]
A joint effort between the Centers for Disease Control and Prevention, the Infectious Disease Society of America, and the American Society of Transplantation and Cellular Therapy established guidelines for the prevention of infections after HSCT.[7] Approaches include preventive or prophylactic antivirals, antifungals, and antibiotics; escalation to heightened empiric therapy for signs of infection; and continued careful monitoring through the full duration of the immunocompromised period after HSCT.
Prophylaxis against fungal infections is standard during the first several months after transplant and may be considered for patients with chronic GVHD who are at high risk of fungal infection. Antifungal prophylaxis must be tailored to the patient's underlying immune status. Pneumocystis infections can occur in all patients after bone marrow transplants, and prophylaxis is mandatory.[6]; [8][Level of evidence C1]
After HSCT, viral infections can be a major source of mortality, especially after T-cell–depleted or cord blood procedures. Types of viral infections include the following:
- Cytomegalovirus (CMV). CMV infection has been a major cause of mortality in the past, but today, effective drugs to treat CMV are available. In addition, preventive strategies, including quantitative polymerase chain reaction (PCR) monitoring followed by preemptive therapy with ganciclovir, have been developed. In addition, the U.S. Food and Drug Administration (FDA) approved letermovir for CMV prophylaxis in adults. There is solid experience using letermovir in children aged 12 years and older and emerging experience in children younger than 12 years.[9,10]
- Epstein-Barr virus (EBV). EBV rarely causes lymphoproliferative disease and is generally associated with intensive, multidrug GVHD therapy or T-cell–depleted HSCT.
- Adenovirus. Adenovirus infection is a major issue in T-cell–depleted transplant, and monitoring by quantitative blood PCR followed by therapy with cidofovir or brincidofovir (available through a compassionate-use protocol) has led to a major decrease in morbidity.[11]
- Other. Other viruses have been implicated in hemorrhagic cystitis (BK virus), encephalitis and poor count recovery (human herpes virus 6), and other clinical issues.[6] One study suggested that high BK viral loads early after transplant (4–7 weeks) may be associated with long-term decreases in glomerular filtration rate.[12]
Careful viral monitoring is essential during high-risk allogeneic procedures.
Late bacterial infections can occur in patients who have central lines or patients with significant chronic GVHD. These patients are susceptible to infection with encapsulated organisms, particularly pneumococcus. Despite reimmunization, these patients can sometimes develop significant infections, and continued prophylaxis is recommended until a serological response to immunizations has been documented. Occasionally, postallogeneic HSCT patients can become functionally asplenic, and antibiotic prophylaxis is recommended. Patients should remain on infection prophylaxis (e.g., Pneumocystis jirovecii pneumonia prophylaxis) until immune recovery. Time to immune recovery varies but ranges from 3 months to 9 months after autologous HSCT, and 9 months to 24 months after allogeneic HSCT without GVHD. Patients with active chronic GVHD may have persistent immunosuppression for years. Many centers monitor T-cell subset recovery after bone marrow transplants as a guide to infection risk.[6]
Vaccination after transplant
International transplant and infectious disease groups have developed specific guidelines for the administration of vaccines after autologous and allogeneic transplants.[6,13,14,15] Comparative studies aimed at defining ideal timing of vaccination after transplant have not been performed, but the vaccine guidelines outlined in Table 2 result in protective titers in most patients who receive vaccinations. These guidelines recommend that autologous transplant recipients receive immunizations beginning at 6 months after stem cell infusion and receive live vaccines 24 months after the transplant. Patients undergoing allogeneic procedures can begin immunizations as soon as 6 months after transplant. However, many groups prefer to wait either until 12 months after the procedure for patients who continue to receive immunosuppressive drugs or until patients are no longer receiving immunosuppressants.
Vaccination recommendations should be reconsidered at times of local endemic or epidemic disease outbreaks. In those settings, earlier vaccination with killed vaccines may be implemented, acknowledging limited host responses. SARS-CoV-2 vaccination recommendations have been included in a recently updated consensus guideline on vaccines after HSCT.[15] Efficacy in gaining protective immunity has been noted with early studies, but given the way the virus has changed over time, the approach to vaccination for SARS-CoV-2 is an ongoing area of study.[16]
Table 2. Vaccination Schedule for Hematopoietic Stem Cell Transplant (HSCT) Recipientsa| Autologous HSCT | 6 Mob | 8 Mob | 12 Mob | 24 Mob |
|---|
| Allogeneic HSCT (if not immunized before 12 mo post-HSCT; start regardless of GVHD status or immunosuppression) | 12 mob(sooner if off immunosuppression) | 14 mob(or 2 mo after first dose) | 18 mob(or 6 mo after first dose) | 24 mob |
|---|
| GVHD = graft-versus-host disease; IM = intramuscular; PO = orally. |
| a Adapted from Tomblyn et al.,[6]Centers for Disease Control and Prevention,[7]and Kumar et al.[17] |
| b Times indicated are times posttransplant (day 0). |
| c Use of Tdap is acceptable if DTap is not available. |
| d Titers may be considered for pediatric patients and patients with GVHD who received immunizations while on immune suppression (minimum 6–8 weeks after last vaccination). |
| e May start as soon as 4 months post-HSCT or sooner for patients with CD4 counts >200/mcL or at any time during an epidemic. If given <6 months after HSCT, may require second dose. Children younger than 9 years require second dose, separated by 1 month. |
| f Consider pre- or postvaccine (at least 6–8 weeks after) titers. |
| g PCV 7 at 24 months only for patients with GVHD; all other patients can get PPV 23. |
| h Pediatric patients should receive two doses at least 1 month apart. |
| Inactivated Vaccines |
| Diphtheria, tetanus, acellular pertussis (DTap) | Xc | Xc | Xc,d | |
| Haemophilus influenzae (Hib) | X | X | Xd | |
| Hepatitis B (HepB) | X | X | Xd | |
| Inactive polio (IPV) | X | X | Xd | |
| Influenza—seasonal injection (IM) | Xe |
| Pneumococcal conjugate (PCV 7, PCV 13) | Xf | X | Xd,f,g | |
| Pneumococcal polysaccharide (PPV 23) | | | Xd,f,g | |
| Live Attenuated Vaccines(contraindicated in patients with active GVHD or on immunosuppression) |
| Measles, mumps, rubella | | | | Xd,h |
| Optional Inactivated Vaccines |
| Hepatitis A | | | Optional | |
| Meningococcal | | | Xd(for high-risk patients) | |
| Optional Live Vaccines(contraindicated in patients with active GVHD or on immunosuppression) |
| Chicken pox (varicella vaccine) | | | | Optional |
| Rabies | | | May be considered at 12–24 mo if exposed |
| Yellow fever, tick-borne encephalitis (TBE), Japanese B encephalitis | | | | For travel in endemic areas |
| Contraindicated Vaccines |
| Intranasal influenza (trivalent live-attenuated influenza vaccine) —household contacts and caregivers should not receive within 2 weeks before contact with HSCT recipient;shingles;bacillus Calmette-Guerin (BCG);oral polio vaccine (OPV);cholera;typhoid vaccine (PO, IM);rotavirus. |
Sinusoidal Obstruction Syndrome/Veno-occlusive Disease (SOS/VOD)
Pathologically, SOS/VOD of the liver is the result of damage to the hepatic sinusoids, resulting in biliary obstruction. This syndrome has been estimated to occur in 15% to 40% of pediatric patients who undergo myeloablative transplants.[18,19]
Risk factors for SOS/VOD include the following:[18,19]
- Use of busulfan (especially before therapeutic pharmacokinetic monitoring).
- Total-body irradiation.
- Serious infection.
- GVHD.
- Preexisting liver dysfunction caused by hepatitis or iron overload.
SOS/VOD is defined clinically by the following:
- Right upper quadrant pain with hepatomegaly.
- Fluid retention (weight gain and ascites).
- Hyperbilirubinemia.
Life-threatening SOS/VOD generally occurs soon after transplant and is characterized by multiorgan system failure.[20] Milder, reversible forms can occur, with full recovery expected. Pediatric patients who have severe SOS/VOD without increased bilirubin have been reported.[21] Therefore, it is important to be vigilant about monitoring patients who have other symptoms without increased bilirubin.
Diagnosis of SOS/VOD
Older definitions of SOS/VOD include the modified Seattle criteria or the Baltimore criteria.
- In the Seattle criteria, at least two of the following must be present by day 20 post HSCT:[22]
- Bilirubin level higher than 2 mg/dL.
- Hepatomegaly or right upper quadrant pain.
- Weight gain (>2%).
- In the Baltimore criteria, a bilirubin level of 2 mg/dL or higher and at least two of the following must be present by day 21 post HSCT:[23]
- Painful hepatomegaly.
- Weight gain (>5%).
- Ascites.
These definitions are inadequate, especially in pediatric practice, as they do not recognize late-onset SOS/VOD or VOD with normal bilirubin levels.
The European Society for Blood and Marrow Transplantation (EBMT) have published revised criteria that are now broadly in use.[24] These criteria recognize late-onset SOS/VOD if proved histologically or have hemodynamic and/or ultrasound evidence of SOS/VOD (hepatomegaly, ascites, and decrease in velocity or reversal of portal flow). They have also included a modification for pediatric patients,[25] with no time limitation for SOS/VOD onset and the presence of two or more of the following:
- Unexplained consumptive and transfusion-refractory thrombocytopenia.
- Otherwise unexplained weight gain on three consecutive days, despite the use of diuretics, or weight gain greater than 5% above baseline value.
- Hepatomegaly above baseline value (best if confirmed by imaging).
- Ascites above baseline value (best if confirmed by imaging).
- Rising bilirubin level from a baseline value on three consecutive days or bilirubin level of 2 mg/dL or higher within 72 hours.
An additional modification of the diagnostic algorithm (Cairo/Cooke criteria) has been proposed, which allows for flexibility with symptoms in unusual situations.[26] The EBMT and Cairo/Cooke criteria have not been prospectively validated in clinical trials.
Prevention and treatment of SOS/VOD
Approaches to both prevention and treatment with agents such as heparin, protein C, and antithrombin III have been studied, with mixed results.[27] One small, retrospective, single-center study showed a benefit from corticosteroid therapy, but further validation is needed.[28]
Another agent with demonstrated activity is defibrotide, a mixture of oligonucleotides with antithrombotic and fibrinolytic effects on microvascular endothelium. Studies of defibrotide have shown the following:
- Decreased mortality in patients who were treated with defibrotide for severe SOS/VOD, compared with historical controls.[29,30,31,32]; [33][Level of evidence C1]
- Decreased SOS/VOD mortality associated with the early initiation of defibrotide treatment soon after diagnostic criteria for SOS/VOD were met.[34][Level of evidence B4]
- Efficacy in decreasing SOS/VOD incidence when used prophylactically.[35][Level of evidence A1] However, a second study was closed due to a lack of efficacy, questioning the validity of prophylactic defibrotide use.[36]
The FDA approved defibrotide for the treatment of patients who have hepatic SOS/VOD with renal or pulmonary dysfunction after HSCT.
The British Society for Blood and Marrow Transplantation (BSBMT) published evidence-guided recommendations for the diagnosis and management of SOS/VOD.[32] They recommend that biopsy be reserved for difficult cases and be performed using the transjugular approach. The BSBMT supports the use of defibrotide for the prevention of SOS/VOD (defibrotide prophylaxis is not currently part of the FDA indication) but maintains there is insufficient data to support the use of prostaglandin E1, pentoxifylline, or antithrombin. For treatment of SOS/VOD, they recommend aggressive fluid balance management, early involvement of critical care and gastroenterology specialists, and the use of defibrotide and possibly methylprednisolone. However, they concluded there is insufficient evidence to support the use of tissue plasminogen activator or N-acetylcysteine.[32,37] The Pediatric Transplantation and Cellular Therapy Consortium, which worked with the Pediatric Acute Lung Injury and Sepsis Investigators, published more detailed consensus recommendations for the diagnosis and management of SOS/VOD in children after HSCT.[38,39,40]
Transplant-Associated Thrombotic Microangiopathy (TA-TMA)
Although TA-TMA clinically mirrors hemolytic uremic syndrome, its causes and clinical course differ from those of other hemolytic uremic syndrome–like diseases. Studies have linked this syndrome with dysregulation of complement pathways.[41] TA-TMA has most frequently been associated with the use of the calcineurin inhibitors tacrolimus and cyclosporine, and it has been noted to occur more frequently when either of these medications is used in combination with sirolimus.[42]
Diagnostic criteria for this syndrome have been updated based on expert consensus opinion and are a modification of criteria published in 2014 (see Table 3).[43,44]
Table 3. TMA Harmonization Panel Consensus Recommended Diagnostic Criteria, Modified Jodele Criteriaa| Biopsy-proven disease (kidney or GI) OR |
|---|
| Clinical criteria: Must meet ≥4 of the following 7 criteria within 14 days at 2 consecutive time points |
|---|
| AIHA = autoimmune hemolytic anemia; BP = blood pressure; GI = gastrointestinal; LDH = lactate dehydrogenase; pRBCs = packed red blood cells; PRCA = pure red cell aplasia; rUPCR = random urine protein to creatinine ratio; ULN = upper limit of normal. |
| a Reprinted with permission from Schoettler et al., which is available under the Creative Commons CC-BY-NC-ND license.[43] |
| b Indicates clarification from published Jodele et al. criteria.[45] |
| Anemiab | Defined as one of the following: |
| 1. Failure to achieve transfusion independence for pRBCs despite evidence of neutrophil engraftment |
| 2. Hemoglobin decline from patient's baseline by 1 g/dL |
| 3. New onset of transfusion dependence |
| Rule out other causes of anemia, such as AIHA and PRCA |
| Thrombocytopeniab | Defined as one of the following: |
| 1. Failure to achieve platelet engraftment |
| 2. Higher than expected platelet transfusion needs |
| 3. Refractoriness to platelet transfusion |
| 4. 50% reduction or greater in baseline platelet count after full platelet engraftment |
| Elevated LDH | >ULN for age |
| Schistocytes | Present |
| Hypertension | >99th percentile for age (<18 y), or systolic BP ≥140 mm Hg or diastolic BP ≥90 mm Hg (≥18 y) |
| Elevated sC5b-9 | ≥ULN |
| Proteinuria | ≥1 mg/mg rUPCR |
Evidence (impact of TA-TMA on HSCT outcomes):
- A multicenter study of TA-TMA in pediatric patients used the following definition of TA-TMA:[46]
- Histological evidence of TA-TMA, or
- Presence of at least four of the following laboratory and clinical markers diagnostic for TA-TMA:
- Lactate dehydrogenase (LDH) levels above reference value for age.
- Schistocytes on peripheral blood smear.
- De novo thrombocytopenia or requirement for platelet transfusions.
- De novo anemia or requirement for red blood cell transfusions.
- Hypertension greater than 99% for age (aged <18 years) or 140/90 mm Hg (aged ≥18 years) requiring ≥2 antihypertensive agents.
- Proteinuria ≥30 mg/dL on random urine analysis twice or random urine protein to creatinine ratio >1 mg/mg.
- Terminal complement activation: Elevated plasma sC5b-9 above normal limit (≥244 ng/mL).
- This study demonstrated the following results:
- In 614 sequential patients who underwent allogeneic or autologous HSCT, 19% of allogeneic recipients and 10% of autologous recipients developed TA-TMA.
- Patients who developed TA-TMA had increased rates of acute GVHD and steroid-refractory GVHD, intensive care unit admission, invasive ventilation, pericardial effusions, pulmonary hypertension, dialysis or continuous renal replacement therapy, acute kidney injury, and VOD.
- In patients who underwent allogeneic HSCT, treatment-related mortality during the first 6 months was significantly higher in patients with TA-TMA than in those without TA-TMA (20% vs. 3%; P ≤ .0001).
- In patients who underwent autologous HSCT, the overall survival (OS) rate during the first 6 months was significantly lower in patients with TA-TMA than in those without TA-TMA (79% vs. 98%; P = .001).
Treatment of TA-TMA
Treatment for TA-TMA includes the following:
- Cessation of the calcineurin inhibitor and substitution with other immune suppressants, if necessary.
- Careful management of hypertension and renal damage by dialysis, if necessary.
Prognosis for normal kidney function when disease is caused by calcineurin inhibitors alone is generally poor. However, most TA-TMA that is associated with the combination of a calcineurin inhibitor and sirolimus has been reversed after sirolimus is discontinued, and in some cases, after both medications are stopped.[42]
Some evidence suggests a role for complement modulation (c5, eculizumab therapy) in preserving renal function. Further assessment of the role of this medication in treating this complication is ongoing.[47,48,49] Although there are no randomized trials that used eculizumab to treat TA-TMA, there are published data from retrospective institutional and multicenter studies and one prospective trial. Historically, the 1-year survival rate for untreated patients with high-risk TA-TMA was about 20%.[50] Two retrospective studies that examined use of eculizumab showed survival that was better than historical controls. A single-center study showed a 1-year OS rate of 66%,[50] and a multicenter study reported a 6-month OS rate of 47% with eculizumab treatment.[51]
Evidence (treatment of high-risk TA-TMA with eculizumab):
A prospective multicenter trial enrolled 21 patients with high-risk TA-TMA and multisystem organ dysfunction. The eculizumab dosing regimen included intensive loading, induction, and maintenance phases for up to 24 weeks of therapy.[44]
- The primary outcome was met, with an OS rate of 71% at 6 months after HSCT (vs. 18% for untreated historical controls; P < .0001).
- Eleven of fifteen survivors (73%) had fully recovered organ function at the time of reporting.
Idiopathic Pneumonia Syndrome (IPS)
IPS is characterized by diffuse, noninfectious lung injury that occurs between 14 and 90 days after the infusion of donor cells. Possible etiologies include direct toxic effects of conditioning regimens and occult infection leading to secretion of high levels of inflammatory cytokines into the alveoli.[52]
The incidence of IPS appears to be decreasing, possibly because of less intensive preparative regimens, better HLA matching, and better definition of occult infections through PCR testing of blood and bronchioalveolar specimens. Mortality rates of 50% to 70% have been reported.[52] However, these estimates are from the mid-1990s, and outcomes may have improved.
Diagnostic criteria include the following signs and symptoms in the absence of documented infectious organisms:[53]
- Pneumonia.
- Evidence of nonlobar radiographic infiltrates.
- Abnormal pulmonary function.
Early assessment by bronchioalveolar lavage to rule out infection is important.
Treatment of IPS
The traditional therapy for IPS has been high-dose methylprednisolone and pulmonary support.
Etanercept is a soluble fusion protein that joins the extracellular ligand-binding domain of the tumor necrosis factor (TNF)–alpha receptor to the Fc region of the immunoglobulin G1 antibody. It acts by blocking TNF-alpha signaling. The addition of etanercept to steroid therapies has shown promising short-term outcomes (extubation, improved short-term survival) in single-center studies.[54] A large phase II trial of this approach in pediatric patients showed promising results, with OS rates of 89% at 1 month and 63% at 12 months.[55]
Autoimmune Cytopenias (AIC)
AIC after allogeneic HSCT can be restricted to one cell lineage (e.g., autoimmune hemolytic anemia), two cell lineages, or three cell lineages. Most data about AIC in pediatric patients after HSCT are reported from single-center experiences, with the number of cases ranging from 20 to 30 over a 10- to 20-year period.[56,57,58] The incidence of AIC is about 5% after allogeneic HSCT. Risk factors for developing AIC seem to be age younger than 10 years and having a nonmalignant disease as an HSCT indication. At least one study has identified use of serotherapy, use of cord blood as the donor source, and severe GVHD as risk factors, but this finding has not been confirmed in other studies. One study demonstrated that patients who develop AIC have inferior outcomes compared with patients who did not develop AIC.[58] However, other studies did not demonstrate an inferior outcome.[56,57]
The National Institutes of Health task force on chronic GVHD has recognized AIC as a possible atypical feature of chronic GVHD (although they may be distinct pathologically).[59] This group has created standardized diagnostic criteria and a proposed prospective study of this complication.
Treatment of AIC
The most common first-line therapy for AIC has been corticosteroids.[56,57,58] Additional immunosuppression or B-cell targeting monoclonal antibodies have been used and produced good responses. Intravenous immunoglobulin is used frequently as adjunct treatment for AIC and/or immunoglobulin replacement.[60] Some clinicians have used bortezomib or daratumumab as third-line agents, with responses noted.[61]
Epstein-Barr Virus (EBV)–Associated Lymphoproliferative Disorder
After HSCT, EBV infection incidence increases through childhood, from approximately 40% in children aged 4 years to more than 80% in teenagers. Patients with a history of previous EBV infection are at risk of EBV reactivation when undergoing HSCT procedures that result in intense, prolonged lymphopenia (T-cell–depleted procedures, use of antithymocyte globulin [ATG] or alemtuzumab, and, to a lesser degree, use of cord blood).[62,63,64]
Features of EBV reactivation can vary, from an isolated increase in EBV titers in the bloodstream as measured by PCR to an aggressive monoclonal disease with marked lymphadenopathy presenting as lymphoma (lymphoproliferative disorder).
Treatment of EBV-associated lymphoproliferative disorder
Isolated bloodstream reactivation of EBV can improve in some cases without therapy as immune function improves. However, lymphoproliferative disorder requires more aggressive therapy.
Treatment of EBV-associated lymphoproliferative disorder involves decreasing immune suppression and treatment with chemotherapy agents such as cyclophosphamide. CD20-positive EBV-associated lymphoproliferative disorder and EBV reactivation have been shown to respond to therapy with the CD20 monoclonal antibody therapy rituximab.[65,66,67] In addition, some centers have shown efficacy in treating or preventing this complication with therapeutic or prophylactic EBV-specific cytotoxic T cells.[68,69,70]
Improved understanding of the risk of EBV reactivation, early monitoring, and aggressive therapy have significantly decreased the risk of mortality from this challenging complication.
Acute GVHD
GVHD is the result of immunologic activation of donor lymphocytes targeting major or minor HLA disparities in the tissues of a recipient.[71] Acute GVHD usually occurs within the first 3 months posttransplant, although delayed acute GVHD has been noted with reduced-intensity conditioning and nonmyeloablative approaches, where achieving a high level of full donor chimerism is sometimes delayed.
Typically, acute GVHD presents with at least one of the following:
- Skin rash.
- Hyperbilirubinemia.
- Secretory diarrhea.
Acute GVHD is classified by staging the severity of skin, liver, and gastrointestinal involvement, and further combining the individual staging of these three areas into an overall grade that is prognostically significant (see Tables 4 and 5).[72] Patients with grade III or grade IV acute GVHD are at higher risk of mortality, generally resulting from organ system damage caused by infections or progressive acute GVHD that is sometimes resistant to therapy.
Table 4. Staging of Acute Graft-Versus-Host Disease (GVHD)a| Stage | Skin | Liver (bilirubin)b | GI/Gut (stool output per day)c |
|---|
| ​ | ​ | ​ | Adult | Child |
|---|
| BSA = body surface area; GI = gastrointestinal. |
| a Adapted from Harris et al.[73] |
| b There is no modification of liver staging for other causes of hyperbilirubinemia. |
| c For GI staging: Theadult stool output values should be used for patients weighing >50 kg. Use 3-day averages for GI staging based on stool output. If stool and urine are mixed, stool output is presumed to be 50% of total stool/urine mix. |
| d If results of colon or rectal biopsy are positive but stool output is <500 mL/day (<10 mL/kg/day), then consider as GI stage 0. |
| e For stage 4 GI: the termsevere abdominal pain will be defined as having both (a) pain control requiring treatment with opioids or an increased dose in ongoing opioid use and (b) pain that significantly impacts performance status, as determined by the treating physician. |
| 0 | No GVHD rash | <2 mg/dL | <500 mL or <3 episodes/day | <10 mL/kg or <4 episodes/day |
| 1 | Maculopapular rash <25% BSA | 2–3 mg/dL | 500–999 mLd or 3–4 episodes/day | 10–19.9 mL/kg or 4–6 episodes/day; persistent nausea, vomiting, or anorexia, with a positive result from upper GI biopsy |
| 2 | Maculopapular rash 25%–50% BSA | 3.1–6 mg/dL | 1,000–1,500 mL or 5–7 episodes/day | 20–30 mL/kg or 7–10 episodes/day |
| 3 | Maculopapular rash >50% BSA | 6.1–15 mg/dL | >1,500 mL or >7 episodes/day | >30 mL/kg or >10 episodes/day |
| 4 | Generalized erythroderma plus bullous formation and desquamation >5% BSA | >15 mg/dL | Severe abdominal paine with or without ileus, or grossly bloody stool (regardless of stool volume) | Severe abdominal paine with or without ileus, or grossly bloody stool (regardless of stool volume) |
Table 5. Overall Clinical Grade (Based on the Highest Stage Obtained)| GI = gastrointestinal. |
| Grade 0: | No stage 1–4 of any organ |
| Grade I: | Stage 1–2 skin and no liver or gut involvement |
| Grade II: | Stage 3 skin and/or stage 1 liver involvement and/or stage 1 GI |
| Grade III: | Stage 0–3 skin, with stage 2–3 liver and/or stage 2–3 GI |
| Grade IV: | Stage 4 skin, liver, or GI involvement |
Because the outcomes of patients with different grades of acute GVHD vary, investigators have sought to more precisely define acute GVHD risk based on serum biomarkers. A study that included both adults and children used a score calculated based on the levels of a combination of three biomarkers (tumor necrosis factor receptor 1 [TNFR1], suppression of tumorigenicity 2 [ST2], and regenerating islet-derived 3-alpha [REG3-alpha]), measured at the onset of acute GVHD. Investigators were able to define patients with low (8%), intermediate (27%), and high (46%, P < .0001) risk of 6-month mortality. The biomarker score was more sensitive and specific for predicting survival than clinical staging.[74] Additional refining of the prediction algorithm showed that measurement of only two biomarkers (ST2 and REG3-alpha) reliably predicted outcome. In addition, after 4 weeks of therapy, changes in the biomarker score were able to further refine prediction of survival outcomes.[75] These findings have led to several studies targeting biomarker high-risk or low-risk subsets of patients with acute GVHD and are influencing clinicians regarding the timing and intensity of acute GVHD therapies.
Prevention and treatment of acute GVHD
Morbidity and mortality from acute GVHD can be reduced through immune suppressive medications given prophylactically or T-cell depletion of grafts, either ex vivo by actual removal of cells from a graft or in vivo with anti–T-lymphocyte antibodies (ATG or anti-CD52 [alemtuzumab]). A newer approach includes administering posttransplant cyclophosphamide on days 3 and 4 after HSCT.[76] While it does not affect stem cells in the graft, cyclophosphamide eliminates or reduces the function or proliferation of alloreactive T cells,[77] markedly decreasing rates of both acute and chronic GVHD.
Complete elimination of acute GVHD with intense T-cell depletion has generally resulted in increased relapse, more infectious morbidity, and increased EBV-associated lymphoproliferative disorder. Because of this result, most HSCT GVHD prophylaxis approaches try to balance risk by giving sufficient immune suppression to prevent severe acute GVHD but not completely remove GVHD risk.
GVHD prophylaxis approaches
GVHD prophylaxis has evolved, from one approach for all donor types to specific and varied approaches tailored to the following factors:
- Stem cell sources. For example, using higher intensity prophylaxis for mismatched bone marrow/peripheral blood stem cell (PBSC) HSCT, compared with low intensity/early immune tapering for matched-sibling bone marrow HSCT.[78]
- Clinical situations. For example, using planned early tapering of prophylaxis for high-risk disease to stimulate the graft-versus-leukemia (GVL) effect.[78]
- Intensity of the HSCT procedure. For example, using less intense prophylaxis for reduced-intensity regimens to increase the GVL effect because of the absence of myeloablation for disease control.
Because of these factors, it is best to consider the combination of preparative regimen, GVHD prophylaxis, and stem cell source as a unit because survival and toxicity outcomes vary if any of these three elements change.
GVHD prophylaxis for matched-sibling HSCT
The most commonly used GVHD prophylaxis approaches in pediatrics for matched-sibling HSCT consist of a calcineurin inhibitor (cyclosporine or tacrolimus), either as a single agent or in combination with methotrexate.[79,80] Doses of methotrexate are often lower for matched-sibling HSCT than for unrelated-donor HSCT. Many centers choose to give prophylaxis on three, rather than four, days after HSCT (days 1, 3, 6, and maybe 11). A large Children's Oncology Group study had good results tapering the calcineurin inhibitor on day 42 and discontinuing it by day 96 when using matched-sibling HSCT,[81] but the traditional taper starts on day 100 posttransplant and continues over 3 months when using unrelated bone marrow/PBSC and cord blood HSCT.
A calcineurin inhibitor in combination with mycophenolate mofetil has also been used with matched-sibling HSCT, especially when reduced-intensity conditioning approaches are used.[82] Posttransplant cyclophosphamide is effective in adult matched-sibling HSCT,[83] but has generally been given with PBSCs in combination with reduced-intensity conditioning approaches. Limited data address the use of posttransplant cyclophosphamide in children undergoing matched-sibling HSCT using bone marrow as a stem cell source,[84] but outcomes appear similar to standard approaches.
GVHD prophylaxis for matched unrelated-donor HSCT
A calcineurin inhibitor in combination with methotrexate (10 mg/m2 for four doses) has been a standard approach that leads to excellent outcomes.[81] However, more recent studies have also shown excellent outcomes with posttransplant cyclophosphamide.[85,86,87]
A number of studies have assessed the role of ATG or alemtuzumab (both considered serotherapy, antibodies that deplete T cells) in improving outcomes after unrelated-donor bone marrow transplant.[88,89,90] Serotherapy includes anti–T-cell approaches (equine ATG and rabbit ATG against either a human T-cell leukemia line [ATG-Fresenius] or human thymocytes [thymoglobulin]) and anti-CD52 antibodies (alemtuzumab). Most centers use serotherapy after HSCT for nonmalignant diseases where GVL is not as important, but the results have varied when treating malignancies. Reasonable evidence suggests that when unrelated-donor PBSCs are used, serotherapy may be beneficial. Studies in children who received unrelated-donor bone marrow HSCT have shown that targeting rabbit ATG in a pharmacokinetic-dependent model leads to faster immune recovery and better outcomes.[91,92]
A combined adult and pediatric study compared abatacept (T-cell costimulatory blocker) plus a calcineurin inhibitor/methotrexate with placebo plus a calcineurin inhibitor/methotrexate. Patients who received abatacept had improved rates of grades 2 to 4 acute GVHD and severe GVHD-free survival.[93]
GVHD prophylaxis for mismatched unrelated-donor HSCT
Use of a calcineurin inhibitor/methotrexate for mismatched unrelated-donor HSCT has led to higher rates of severe GVHD and lower rates of survival. This outcome is partially mitigated by the use of serotherapy in combination with a calcineurin inhibitor/methotrexate. Using this combined approach, the International BFM Study Group has considered the use of a single-antigen mismatched donor (7/8 or 9/10) to be a well-matched donor, with outcomes similar to those with matched unrelated donors.[94]
A prospective trial in mismatched unrelated-donor recipients added abatacept to a calcineurin inhibitor/methotrexate regimen. The study showed a marked improvement in severe acute GVHD and OS, compared with a Center for International Blood and Marrow Transplant Research control trial that used a calcineurin inhibitor/methotrexate alone.[93] Posttransplant cyclophosphamide approaches have also led to improvements in outcomes when mismatched unrelated-donor HSCT have been used. A key trial run by the National Marrow Donor Program tested posttransplant cyclophosphamide using single and multiply mismatched unrelated donors. The results showed impressive decreases in acute and chronic GVHD and improved event-free survival and OS.[95] While these new approaches have revived interest in mismatched unrelated-donor use, prospective trials comparing mismatched unrelated donors using these new approaches with unrelated cord blood or haploidentical stem cell sources have not been performed.
GVHD prophylaxis for unrelated-donor cord blood HSCT
A calcineurin inhibitor/methotrexate and cyclosporine/prednisone regimen has been used as GVHD prophylaxis in cord blood HSCT. However, a number of studies in pediatric patients have documented better survival and GVHD outcomes using a calcineurin inhibitor/mycophenolate mofetil combination.[95,96,97] Although serotherapy has often been used for cord blood HSCT, especially in the context of nonmalignant indications, there is good evidence of improved outcomes and faster immune reconstitution without serotherapy.[98,99] Targeted dosing of serotherapy shows improved outcomes when used during cord blood HSCT, but a comparison of targeted serotherapy with no serotherapy has not been performed.
GVHD prophylaxis for haploidentical-donor HSCT
Early approaches using various intensities of GVHD prophylaxis and different types of T-cell depletion led to relatively poor rates of survival and high rates of GVHD when haploidentical donors were used.[100] The approach has changed dramatically with the use of posttransplant cyclophosphamide, both for bone marrow and PBSCs, leading to outcomes comparable to those for fully matched unrelated donors.[85]
The other widely used approach to haploidentical HSCT in pediatrics is T-cell receptor (TCR) alpha beta/CD19 depletion. Using this process, several pediatric groups have demonstrated outcomes similar to those for fully matched stem cell sources, with low rates of GVHD.[101,102,103] Studies that compare this method with posttransplant cyclophosphamide or other stem cell sources have not been performed. Other approaches to T-cell depletion (e.g., CD34+ isolation,[104] CD45RA depletion [105]) are used by some centers, but comparative studies with other approaches are not available.
Nutritional approaches to prevent GVHD
Other nonimmune approaches to prevent GVHD are emerging. In a double-blind randomized study, patients with low vitamin A levels received either one pretransplant dose of vitamin A or a placebo. Patients who received vitamin A had statistically less acute GVHD (grades II to IV), acute gastrointestinal GVHD, and chronic GVHD.[106]
Steroid-refractory acute GVHD
When significant acute GVHD occurs, first-line therapy is generally methylprednisolone.[107] Patients with acute GVHD who are resistant to this therapy have a poor prognosis. However, many patients respond to second-line agents (e.g., mycophenolate mofetil, infliximab, pentostatin, sirolimus, or extracorporeal photopheresis).[108] Ruxolitinib was approved in 2019 for the treatment of children aged 12 years and older with steroid-refractory acute GVHD, with an overall response rate of 55% and a complete response rate of 27% at day 28 after initiation of therapy. Comparative trials of these agents have not been performed, so a best option for steroid-refractory GVHD has not been identified.[109,110]
References:
- Antin JH: Immune reconstitution: the major barrier to successful stem cell transplantation. Biol Blood Marrow Transplant 11 (2 Suppl 2): 43-5, 2005.
- Fry TJ, Mackall CL: Immune reconstitution following hematopoietic progenitor cell transplantation: challenges for the future. Bone Marrow Transplant 35 (Suppl 1): S53-7, 2005.
- Wingard JR, Majhail NS, Brazauskas R, et al.: Long-term survival and late deaths after allogeneic hematopoietic cell transplantation. J Clin Oncol 29 (16): 2230-9, 2011.
- Bunin N, Small T, Szabolcs P, et al.: NCI, NHLBI/PBMTC first international conference on late effects after pediatric hematopoietic cell transplantation: persistent immune deficiency in pediatric transplant survivors. Biol Blood Marrow Transplant 18 (1): 6-15, 2012.
- Burik JH, Freifeld AG: Infection in the severely immunocompromised patient. In: Abeloff MD, Armitage JO, Niederhuber JE, et al.: Clinical Oncology. 3rd ed. Elsevier, Churchill Livingstone, 2004, pp 941-56.
- Tomblyn M, Chiller T, Einsele H, et al.: Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant 15 (10): 1143-238, 2009.
- Centers for Disease Control and Prevention, Infectious Disease Society of America, American Society of Blood and Marrow Transplantation: Guidelines for preventing opportunistic infections among hematopoietic stem cell transplant recipients. MMWR Recomm Rep 49 (RR-10): 1-125, CE1-7, 2000.
- Levy ER, Musick L, Zinter MS, et al.: Safe and Effective Prophylaxis with Bimonthly Intravenous Pentamidine in the Pediatric Hematopoietic Stem Cell Transplant Population. Pediatr Infect Dis J 35 (2): 135-41, 2016.
- Russo D, Schmitt M, Pilorge S, et al.: Efficacy and safety of extended duration letermovir prophylaxis in recipients of haematopoietic stem-cell transplantation at risk of cytomegalovirus infection: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol 11 (2): e127-e135, 2024.
- Galaverna F, Baccelli F, Zama D, et al.: Letermovir for Cytomegalovirus infection in pediatric patients undergoing allogenic hematopoietic stem cell transplantation: a real-life study by the Infectious Diseases Working Group of Italian Association of Pediatric Hematology-Oncology (AIEOP). Bone Marrow Transplant 59 (4): 505-512, 2024.
- Hiwarkar P, Amrolia P, Sivaprakasam P, et al.: Brincidofovir is highly efficacious in controlling adenoviremia in pediatric recipients of hematopoietic cell transplant. Blood 129 (14): 2033-2037, 2017.
- Wychera C, Imlay HN, Duke ER, et al.: BK Viremia and Changes in Estimated Glomerular Filtration Rate in Children and Young Adults after Hematopoietic Cell Transplantation. Transplant Cell Ther 29 (3): 187.e1-187.e8, 2023.
- Rubin LG, Levin MJ, Ljungman P, et al.: 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 58 (3): e44-100, 2014.
- Cordonnier C, Einarsdottir S, Cesaro S, et al.: Vaccination of haemopoietic stem cell transplant recipients: guidelines of the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect Dis 19 (6): e200-e212, 2019.
- Miller P, Patel SR, Skinner R, et al.: Joint consensus statement on the vaccination of adult and paediatric haematopoietic stem cell transplant recipients: Prepared on behalf of the British society of blood and marrow transplantation and cellular therapy (BSBMTCT), the Children's cancer and Leukaemia Group (CCLG), and British Infection Association (BIA). J Infect 86 (1): 1-8, 2023.
- Hill JA, Martens MJ, Young JH, et al.: SARS-CoV-2 vaccination in the first year after allogeneic hematopoietic cell transplant: a prospective, multicentre, observational study. EClinicalMedicine 59: 101983, 2023.
- Kumar D, Chen MH, Welsh B, et al.: A randomized, double-blind trial of pneumococcal vaccination in adult allogeneic stem cell transplant donors and recipients. Clin Infect Dis 45 (12): 1576-82, 2007.
- Reiss U, Cowan M, McMillan A, et al.: Hepatic venoocclusive disease in blood and bone marrow transplantation in children and young adults: incidence, risk factors, and outcome in a cohort of 241 patients. J Pediatr Hematol Oncol 24 (9): 746-50, 2002.
- Cesaro S, Pillon M, Talenti E, et al.: A prospective survey on incidence, risk factors and therapy of hepatic veno-occlusive disease in children after hematopoietic stem cell transplantation. Haematologica 90 (10): 1396-404, 2005.
- Bearman SI: The syndrome of hepatic veno-occlusive disease after marrow transplantation. Blood 85 (11): 3005-20, 1995.
- Myers KC, Dandoy C, El-Bietar J, et al.: Veno-occlusive disease of the liver in the absence of elevation in bilirubin in pediatric patients after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 21 (2): 379-81, 2015.
- Carreras E, Dufour C, Mohty M, et al., eds.: The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. 7th edition. Cham (CH): Springer, 2019. Available online. Last accessed May 7, 2024.
- Jones RJ, Lee KS, Beschorner WE, et al.: Venoocclusive disease of the liver following bone marrow transplantation. Transplantation 44 (6): 778-83, 1987.
- Mohty M, Malard F, Abecassis M, et al.: Revised diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant 51 (7): 906-12, 2016.
- Corbacioglu S, Carreras E, Ansari M, et al.: Diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in pediatric patients: a new classification from the European society for blood and marrow transplantation. Bone Marrow Transplant 53 (2): 138-145, 2018.
- Cairo MS, Cooke KR, Lazarus HM, et al.: Modified diagnostic criteria, grading classification and newly elucidated pathophysiology of hepatic SOS/VOD after haematopoietic cell transplantation. Br J Haematol 190 (6): 822-836, 2020.
- Ruutu T, Eriksson B, Remes K, et al.: Ursodeoxycholic acid for the prevention of hepatic complications in allogeneic stem cell transplantation. Blood 100 (6): 1977-83, 2002.
- Myers KC, Lawrence J, Marsh RA, et al.: High-dose methylprednisolone for veno-occlusive disease of the liver in pediatric hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant 19 (3): 500-3, 2013.
- Richardson PG, Murakami C, Jin Z, et al.: Multi-institutional use of defibrotide in 88 patients after stem cell transplantation with severe veno-occlusive disease and multisystem organ failure: response without significant toxicity in a high-risk population and factors predictive of outcome. Blood 100 (13): 4337-43, 2002.
- Corbacioglu S, Kernan N, Lehmann L, et al.: Defibrotide for the treatment of hepatic veno-occlusive disease in children after hematopoietic stem cell transplantation. Expert Rev Hematol 5 (3): 291-302, 2012.
- Richardson PG, Soiffer RJ, Antin JH, et al.: Defibrotide for the treatment of severe hepatic veno-occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose-finding trial. Biol Blood Marrow Transplant 16 (7): 1005-17, 2010.
- Dignan FL, Wynn RF, Hadzic N, et al.: BCSH/BSBMT guideline: diagnosis and management of veno-occlusive disease (sinusoidal obstruction syndrome) following haematopoietic stem cell transplantation. Br J Haematol 163 (4): 444-57, 2013.
- Strouse C, Richardson P, Prentice G, et al.: Defibrotide for Treatment of Severe Veno-Occlusive Disease in Pediatrics and Adults: An Exploratory Analysis Using Data from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant 22 (7): 1306-1312, 2016.
- Richardson PG, Smith AR, Triplett BM, et al.: Earlier defibrotide initiation post-diagnosis of veno-occlusive disease/sinusoidal obstruction syndrome improves Day +100 survival following haematopoietic stem cell transplantation. Br J Haematol 178 (1): 112-118, 2017.
- Corbacioglu S, Cesaro S, Faraci M, et al.: Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet 379 (9823): 1301-9, 2012.
- Grupp SA, Corbacioglu S, Kang HJ, et al.: Defibrotide plus best standard of care compared with best standard of care alone for the prevention of sinusoidal obstruction syndrome (HARMONY): a randomised, multicentre, phase 3 trial. Lancet Haematol 10 (5): e333-e345, 2023.
- Ruutu T, Juvonen E, Remberger M, et al.: Improved survival with ursodeoxycholic acid prophylaxis in allogeneic stem cell transplantation: long-term follow-up of a randomized study. Biol Blood Marrow Transplant 20 (1): 135-8, 2014.
- Bajwa RPS, Mahadeo KM, Taragin BH, et al.: Consensus Report by Pediatric Acute Lung Injury and Sepsis Investigators and Pediatric Blood and Marrow Transplantation Consortium Joint Working Committees: Supportive Care Guidelines for Management of Veno-Occlusive Disease in Children and Adolescents, Part 1: Focus on Investigations, Prophylaxis, and Specific Treatment. Biol Blood Marrow Transplant 23 (11): 1817-1825, 2017.
- Mahadeo KM, McArthur J, Adams RH, et al.: Consensus Report by the Pediatric Acute Lung Injury and Sepsis Investigators and Pediatric Blood and Marrow Transplant Consortium Joint Working Committees on Supportive Care Guidelines for Management of Veno-Occlusive Disease in Children and Adolescents: Part 2-Focus on Ascites, Fluid and Electrolytes, Renal, and Transfusion Issues. Biol Blood Marrow Transplant 23 (12): 2023-2033, 2017.
- Ovchinsky N, Frazier W, Auletta JJ, et al.: Consensus Report by the Pediatric Acute Lung Injury and Sepsis Investigators and Pediatric Blood and Marrow Transplantation Consortium Joint Working Committees on Supportive Care Guidelines for Management of Veno-Occlusive Disease in Children and Adolescents, Part 3: Focus on Cardiorespiratory Dysfunction, Infections, Liver Dysfunction, and Delirium. Biol Blood Marrow Transplant 24 (2): 207-218, 2018.
- Jodele S, Licht C, Goebel J, et al.: Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood 122 (12): 2003-7, 2013.
- Cutler C, Henry NL, Magee C, et al.: Sirolimus and thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 11 (7): 551-7, 2005.
- Schoettler ML, Carreras E, Cho B, et al.: Harmonizing Definitions for Diagnostic Criteria and Prognostic Assessment of Transplantation-Associated Thrombotic Microangiopathy: A Report on Behalf of the European Society for Blood and Marrow Transplantation, American Society for Transplantation and Cellular Therapy, Asia-Pacific Blood and Marrow Transplantation Group, and Center for International Blood and Marrow Transplant Research. Transplant Cell Ther 29 (3): 151-163, 2023.
- Jodele S, Dandoy CE, Aguayo-Hiraldo P, et al.: A prospective multi-institutional study of eculizumab to treat high-risk stem cell transplantation-associated TMA. Blood 143 (12): 1112-1123, 2024.
- Jodele S, Davies SM, Lane A, et al.: Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood 124 (4): 645-53, 2014.
- Dandoy CE, Rotz S, Alonso PB, et al.: A pragmatic multi-institutional approach to understanding transplant-associated thrombotic microangiopathy after stem cell transplant. Blood Adv 5 (1): 1-11, 2021.
- Jodele S, Fukuda T, Vinks A, et al.: Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transplant 20 (4): 518-25, 2014.
- Jodele S, Fukuda T, Mizuno K, et al.: Variable Eculizumab Clearance Requires Pharmacodynamic Monitoring to Optimize Therapy for Thrombotic Microangiopathy after Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant 22 (2): 307-315, 2016.
- Schoettler M, Lehmann L, Li A, et al.: Thrombotic Microangiopathy Following Pediatric Autologous Hematopoietic Cell Transplantation: A Report of Significant End-Organ Dysfunction in Eculizumab-Treated Survivors. Biol Blood Marrow Transplant 25 (5): e163-e168, 2019.
- Jodele S, Dandoy CE, Lane A, et al.: Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood 135 (13): 1049-1057, 2020.
- Svec P, Elfeky R, Galimard JE, et al.: Use of eculizumab in children with allogeneic haematopoietic stem cell transplantation associated thrombotic microangiopathy - a multicentre retrospective PDWP and IEWP EBMT study. Bone Marrow Transplant 58 (2): 129-141, 2023.
- Kantrow SP, Hackman RC, Boeckh M, et al.: Idiopathic pneumonia syndrome: changing spectrum of lung injury after marrow transplantation. Transplantation 63 (8): 1079-86, 1997.
- Clark JG, Hansen JA, Hertz MI, et al.: NHLBI workshop summary. Idiopathic pneumonia syndrome after bone marrow transplantation. Am Rev Respir Dis 147 (6 Pt 1): 1601-6, 1993.
- Yanik GA, Ho VT, Levine JE, et al.: The impact of soluble tumor necrosis factor receptor etanercept on the treatment of idiopathic pneumonia syndrome after allogeneic hematopoietic stem cell transplantation. Blood 112 (8): 3073-81, 2008.
- Yanik GA, Grupp SA, Pulsipher MA, et al.: TNF-receptor inhibitor therapy for the treatment of children with idiopathic pneumonia syndrome. A joint Pediatric Blood and Marrow Transplant Consortium and Children's Oncology Group Study (ASCT0521). Biol Blood Marrow Transplant 21 (1): 67-73, 2015.
- Szanto CL, Langenhorst J, de Koning C, et al.: Predictors for Autoimmune Cytopenias after Allogeneic Hematopoietic Cell Transplantation in Children. Biol Blood Marrow Transplant 26 (1): 114-122, 2020.
- Koo J, Giller RH, Quinones R, et al.: Autoimmune cytopenias following allogeneic hematopoietic stem cell transplant in pediatric patients: Response to therapy and late effects. Pediatr Blood Cancer 67 (9): e28591, 2020.
- O'Brien TA, Eastlund T, Peters C, et al.: Autoimmune haemolytic anaemia complicating haematopoietic cell transplantation in paediatric patients: high incidence and significant mortality in unrelated donor transplants for non-malignant diseases. Br J Haematol 127 (1): 67-75, 2004.
- Cuvelier GDE, Schoettler M, Buxbaum NP, et al.: Toward a Better Understanding of the Atypical Features of Chronic Graft-Versus-Host Disease: A Report from the 2020 National Institutes of Health Consensus Project Task Force. Transplant Cell Ther 28 (8): 426-445, 2022.
- Hillier K, Harris EM, Berbert L, et al.: Characteristics and outcomes of autoimmune hemolytic anemia after pediatric allogeneic stem cell transplant. Pediatr Blood Cancer 69 (1): e29410, 2022.
- Even-Or E, Schejter YD, NaserEddin A, et al.: Autoimmune Cytopenias Post Hematopoietic Stem Cell Transplantation in Pediatric Patients With Osteopetrosis and Other Nonmalignant Diseases. Front Immunol 13: 879994, 2022.
- Gerritsen EJ, Stam ED, Hermans J, et al.: Risk factors for developing EBV-related B cell lymphoproliferative disorders (BLPD) after non-HLA-identical BMT in children. Bone Marrow Transplant 18 (2): 377-82, 1996.
- Shapiro RS, McClain K, Frizzera G, et al.: Epstein-Barr virus associated B cell lymphoproliferative disorders following bone marrow transplantation. Blood 71 (5): 1234-43, 1988.
- Brunstein CG, Weisdorf DJ, DeFor T, et al.: Marked increased risk of Epstein-Barr virus-related complications with the addition of antithymocyte globulin to a nonmyeloablative conditioning prior to unrelated umbilical cord blood transplantation. Blood 108 (8): 2874-80, 2006.
- Blaes AH, Cao Q, Wagner JE, et al.: Monitoring and preemptive rituximab therapy for Epstein-Barr virus reactivation after antithymocyte globulin containing nonmyeloablative conditioning for umbilical cord blood transplantation. Biol Blood Marrow Transplant 16 (2): 287-91, 2010.
- Kuehnle I, Huls MH, Liu Z, et al.: CD20 monoclonal antibody (rituximab) for therapy of Epstein-Barr virus lymphoma after hemopoietic stem-cell transplantation. Blood 95 (4): 1502-5, 2000.
- Styczynski J, Gil L, Tridello G, et al.: Response to rituximab-based therapy and risk factor analysis in Epstein Barr Virus-related lymphoproliferative disorder after hematopoietic stem cell transplant in children and adults: a study from the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Clin Infect Dis 57 (6): 794-802, 2013.
- Liu Z, Savoldo B, Huls H, et al.: Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the prevention and treatment of EBV-associated post-transplant lymphomas. Recent Results Cancer Res 159: 123-33, 2002.
- Bollard CM, Heslop HE: T cells for viral infections after allogeneic hematopoietic stem cell transplant. Blood 127 (26): 3331-40, 2016.
- Keller MD, Hanley PJ, Chi YY, et al.: Antiviral cellular therapy for enhancing T-cell reconstitution before or after hematopoietic stem cell transplantation (ACES): a two-arm, open label phase II interventional trial of pediatric patients with risk factor assessment. Nat Commun 15 (1): 3258, 2024.
- Ferrara JL, Levine JE, Reddy P, et al.: Graft-versus-host disease. Lancet 373 (9674): 1550-61, 2009.
- Przepiorka D, Weisdorf D, Martin P, et al.: 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant 15 (6): 825-8, 1995.
- Harris AC, Young R, Devine S, et al.: International, Multicenter Standardization of Acute Graft-versus-Host Disease Clinical Data Collection: A Report from the Mount Sinai Acute GVHD International Consortium. Biol Blood Marrow Transplant 22 (1): 4-10, 2016.
- Levine JE, Braun TM, Harris AC, et al.: A prognostic score for acute graft-versus-host disease based on biomarkers: a multicentre study. Lancet Haematol 2 (1): e21-9, 2015.
- Srinagesh HK, Özbek U, Kapoor U, et al.: The MAGIC algorithm probability is a validated response biomarker of treatment of acute graft-versus-host disease. Blood Adv 3 (23): 4034-4042, 2019.
- Luznik L, O'Donnell PV, Fuchs EJ: Post-transplantation cyclophosphamide for tolerance induction in HLA-haploidentical bone marrow transplantation. Semin Oncol 39 (6): 683-93, 2012.
- Nunes NS, Kanakry CG: Mechanisms of Graft-versus-Host Disease Prevention by Post-transplantation Cyclophosphamide: An Evolving Understanding. Front Immunol 10: 2668, 2019.
- Watkins B, Williams KM: Controversies and expectations for the prevention of GVHD: A biological and clinical perspective. Front Immunol 13: 1057694, 2022.
- Locatelli F, Zecca M, Rondelli R, et al.: Graft versus host disease prophylaxis with low-dose cyclosporine-A reduces the risk of relapse in children with acute leukemia given HLA-identical sibling bone marrow transplantation: results of a randomized trial. Blood 95 (5): 1572-9, 2000.
- Storb R, Deeg HJ, Pepe M, et al.: Methotrexate and cyclosporine versus cyclosporine alone for prophylaxis of graft-versus-host disease in patients given HLA-identical marrow grafts for leukemia: long-term follow-up of a controlled trial. Blood 73 (6): 1729-34, 1989.
- Pulsipher MA, Langholz B, Wall DA, et al.: The addition of sirolimus to tacrolimus/methotrexate GVHD prophylaxis in children with ALL: a phase 3 Children's Oncology Group/Pediatric Blood and Marrow Transplant Consortium trial. Blood 123 (13): 2017-25, 2014.
- Ueda Oshima M, Storer BE, Qiu H, et al.: Long-term Outcomes with Nonmyeloablative HLA-Identical Related Hematopoietic Cell Transplantation Using Tacrolimus and Mycophenolate Mofetil for Graft-versus-Host Disease Prophylaxis. Transplant Cell Ther 27 (2): 163.e1-163.e7, 2021.
- Mehta RS, Saliba RM, Rondon G, et al.: Post-Transplantation Cyclophosphamide Versus Tacrolimus and Methotrexate Graft-Versus-Host Disease Prophylaxis for HLA-Matched Donor Transplantation. Transplant Cell Ther 28 (10): 695.e1-695.e10, 2022.
- Borovkova AS, Paina OV, Semenova EV, et al.: Post-transplant Ñyclophosphamide after matched donor hematopoietic stem cell transplantation in children with acute leukemia. Clin Transplant 38 (1): e15181, 2024.
- Fierro-Pineda JC, Tsai HL, Blackford A, et al.: Prospective PTCTC trial of myeloablative haplo-BMT with posttransplant cyclophosphamide for pediatric acute leukemias. Blood Adv 7 (18): 5639-5648, 2023.
- McCurdy SR, Kanakry CG, Tsai HL, et al.: Development of Grade II Acute Graft-versus-Host Disease Is Associated with Improved Survival after Myeloablative HLA-Matched Bone Marrow Transplantation using Single-Agent Post-Transplant Cyclophosphamide. Biol Blood Marrow Transplant 25 (6): 1128-1135, 2019.
- Kanakry CG, O'Donnell PV, Furlong T, et al.: Multi-institutional study of post-transplantation cyclophosphamide as single-agent graft-versus-host disease prophylaxis after allogeneic bone marrow transplantation using myeloablative busulfan and fludarabine conditioning. J Clin Oncol 32 (31): 3497-505, 2014.
- Soiffer RJ, Kim HT, McGuirk J, et al.: Prospective, Randomized, Double-Blind, Phase III Clinical Trial of Anti-T-Lymphocyte Globulin to Assess Impact on Chronic Graft-Versus-Host Disease-Free Survival in Patients Undergoing HLA-Matched Unrelated Myeloablative Hematopoietic Cell Transplantation. J Clin Oncol 35 (36): 4003-4011, 2017.
- Socié G, Schmoor C, Bethge WA, et al.: Chronic graft-versus-host disease: long-term results from a randomized trial on graft-versus-host disease prophylaxis with or without anti-T-cell globulin ATG-Fresenius. Blood 117 (23): 6375-82, 2011.
- Kröger N, Solano C, Wolschke C, et al.: Antilymphocyte Globulin for Prevention of Chronic Graft-versus-Host Disease. N Engl J Med 374 (1): 43-53, 2016.
- Admiraal R, Nierkens S, Bierings MB, et al.: Individualised dosing of anti-thymocyte globulin in paediatric unrelated allogeneic haematopoietic stem-cell transplantation (PARACHUTE): a single-arm, phase 2 clinical trial. Lancet Haematol 9 (2): e111-e120, 2022.
- Admiraal R, van Kesteren C, Jol-van der Zijde CM, et al.: Association between anti-thymocyte globulin exposure and CD4+ immune reconstitution in paediatric haemopoietic cell transplantation: a multicentre, retrospective pharmacodynamic cohort analysis. Lancet Haematol 2 (5): e194-203, 2015.
- Watkins B, Qayed M, McCracken C, et al.: Phase II Trial of Costimulation Blockade With Abatacept for Prevention of Acute GVHD. J Clin Oncol 39 (17): 1865-1877, 2021.
- Peters C, Schrappe M, von Stackelberg A, et al.: Stem-cell transplantation in children with acute lymphoblastic leukemia: A prospective international multicenter trial comparing sibling donors with matched unrelated donors-The ALL-SCT-BFM-2003 trial. J Clin Oncol 33 (11): 1265-74, 2015.
- Shaw BE, Jimenez-Jimenez AM, Burns LJ, et al.: National Marrow Donor Program-Sponsored Multicenter, Phase II Trial of HLA-Mismatched Unrelated Donor Bone Marrow Transplantation Using Post-Transplant Cyclophosphamide. J Clin Oncol 39 (18): 1971-1982, 2021.
- Wagner JE, Eapen M, Carter S, et al.: One-unit versus two-unit cord-blood transplantation for hematologic cancers. N Engl J Med 371 (18): 1685-94, 2014.
- Eapen M, Kurtzberg J, Zhang MJ, et al.: Umbilical Cord Blood Transplantation in Children with Acute Leukemia: Impact of Conditioning on Transplantation Outcomes. Biol Blood Marrow Transplant 23 (10): 1714-1721, 2017.
- Lindemans CA, Chiesa R, Amrolia PJ, et al.: Impact of thymoglobulin prior to pediatric unrelated umbilical cord blood transplantation on immune reconstitution and clinical outcome. Blood 123 (1): 126-32, 2014.
- Admiraal R, Lindemans CA, van Kesteren C, et al.: Excellent T-cell reconstitution and survival depend on low ATG exposure after pediatric cord blood transplantation. Blood 128 (23): 2734-2741, 2016.
- Pulsipher MA: Haplo is the new black. Blood 124 (5): 675-6, 2014.
- Pulsipher MA, Ahn KW, Bunin NJ, et al.: KIR-favorable TCR-αβ/CD19-depleted haploidentical HCT in children with ALL/AML/MDS: primary analysis of the PTCTC ONC1401 trial. Blood 140 (24): 2556-2572, 2022.
- Bertaina A, Zecca M, Buldini B, et al.: Unrelated donor vs HLA-haploidentical α/β T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood 132 (24): 2594-2607, 2018.
- Merli P, Algeri M, Galaverna F, et al.: TCRαβ/CD19 cell-depleted HLA-haploidentical transplantation to treat pediatric acute leukemia: updated final analysis. Blood 143 (3): 279-289, 2024.
- Mehta PA, Davies SM, Leemhuis T, et al.: Radiation-free, alternative-donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 129 (16): 2308-2315, 2017.
- Bleakley M, Heimfeld S, Loeb KR, et al.: Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest 125 (7): 2677-89, 2015.
- Khandelwal P, Langenberg L, Luebbering N, et al.: A randomized phase 2 trial of oral vitamin A for graft-versus-host disease in children and young adults. Blood 143 (12): 1181-1192, 2024.
- Jacobsohn DA: Acute graft-versus-host disease in children. Bone Marrow Transplant 41 (2): 215-21, 2008.
- Deeg HJ: How I treat refractory acute GVHD. Blood 109 (10): 4119-26, 2007.
- Jagasia M, Perales MA, Schroeder MA, et al.: Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood 135 (20): 1739-1749, 2020.
- Laisne L, Neven B, Dalle JH, et al.: Ruxolitinib in children with steroid-refractory acute graft-versus-host disease: A retrospective multicenter study of the pediatric group of SFGM-TC. Pediatr Blood Cancer 67 (9): e28233, 2020.
Chronic Graft-Versus-Host Disease (GVHD)
Chronic GVHD is a syndrome that can involve a single organ system or several organ systems, with clinical features resembling an autoimmune disease.[1,2] Chronic GVHD is usually first noted 2 to 12 months after hematopoietic stem cell transplant (HSCT). Traditionally, symptoms occurring more than 100 days after HSCT were considered chronic GVHD, and symptoms occurring sooner than 100 days after HSCT were considered acute GVHD. Because some approaches to HSCT can lead to late-onset acute GVHD, and manifestations that are diagnostic for chronic GVHD can occur sooner than 100 days post-HSCT, the following three distinct types of chronic GVHD have been described:
- Classic chronic GVHD: Occurs with diagnostic and/or distinct features of chronic GVHD (see Tables 6–10) after a previous history of resolved acute GVHD.
- Overlap syndrome: An ongoing GVHD process when manifestations diagnostic for chronic GVHD occur while symptoms of acute GVHD persist.
- De novo chronic GVHD: New-onset GVHD generally occurring at least 2 months after transplant, with diagnostic and/or distinct features of chronic GVHD and no history or features of acute GVHD.
Organ Manifestations of Chronic GVHD
The diagnosis of chronic GVHD is based on clinical features (at least one diagnostic clinical sign, e.g., poikiloderma) or distinctive manifestations complemented by relevant tests (e.g., dry eye with positive results of a Schirmer test).[3]
Commonly involved tissues include the skin, eyes, mouth, hair, joints, liver, and gastrointestinal tract. Other tissues such as lungs, nails, muscles, urogenital system, and nervous system may also be involved. Tables 6 to 10 list organ manifestations of chronic GVHD, including a description of findings that are sufficient to establish the diagnosis of chronic GVHD. Biopsies of affected sites may be needed to confirm the diagnosis.[4]
Common skin manifestations include alterations in pigmentation, texture, elasticity, and thickness, with papules, plaques, or follicular changes. Patient-reported symptoms include dry skin, itching, limited mobility, rash, sores, or changes in coloring or texture. Generalized scleroderma may lead to severe joint contractures and debility. Associated hair loss and nail changes are common. Other important symptoms that should be assessed include dry eyes and oral changes such as atrophy, ulcers, and lichen planus. In addition, joint stiffness along with restricted range of motion, weight loss, nausea, difficulty swallowing, and diarrhea should be noted.
Table 6. Chronic Graft-Versus-Host Disease (GVHD) Symptoms in the Skin, Nails, Scalp, and Body Haira| Organ or Site | Diagnosticb | Distinctivec | Other Featuresd | Common (Seen With Both Acute and Chronic GVHD) |
|---|
| a Reprinted fromBiology of Blood and Marrow Transplantation, Volume 11 (Issue 12), Alexandra H. Filipovich, Daniel Weisdorf, Steven Pavletic, Gerard Socie, John R. Wingard, Stephanie J. Lee, Paul Martin, Jason Chien, Donna Przepiorka, Daniel Couriel, Edward W. Cowen, Patricia Dinndorf, Ann Farrell, Robert Hartzman, Jean Henslee-Downey, David Jacobsohn, George McDonald, Barbara Mittleman, J. Douglas Rizzo, Michael Robinson, Mark Schubert, Kirk Schultz, Howard Shulman, Maria Turner, Georgia Vogelsang, Mary E.D. Flowers, National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. Diagnosis and Staging Working Group Report, Pages 945-956, Copyright 2005, with permission from American Society for Blood and Marrow Transplantation and Elsevier.[3] |
| b Sufficient to establish a diagnosis of chronic GVHD. |
| c Seen in chronic GVHD but insufficient alone to establish a diagnosis of chronic GVHD. |
| d Can be acknowledged as part of the chronic GVHD symptomatology if the diagnosis is confirmed. |
| e In all cases, infection, drug effects, malignancy, or other causes must be excluded. |
| f Diagnosis of chronic GVHD requires biopsy or radiology confirmation (or Schirmer test for eyes). |
| Skin | Poikiloderma | Depigmentation | Sweat impairment | Pruritus |
| Lichen planus–like features | Ichthyosis | Erythema |
| Sclerotic features | Keratosis pilaris | Maculopapular rash |
| Morphea-like features | Hypopigmentation |
| Lichen sclerosus–like features | Hyperpigmentation |
|
| Nails | | Dystrophy | | |
| Longitudinal ridging, splitting, or brittle features |
| Onycholysis |
| Pterygium unguis |
| Nail loss (usually symmetric; affects most nails)e |
|
| Scalp and body hair | | New onset of scarring or nonscarring scalp alopecia (after recovery from chemoradiotherapy) | Thinning scalp hair, typically patchy, coarse, or dull (not explained by endocrine or other causes) | |
| Scaling, papulosquamous lesions | Premature gray hair |
Table 7. Chronic Graft-Versus-Host Disease (GVHD) Symptoms in the Mouth and GI Tracta| Organ or Site | Diagnosticb | Distinctivec | Other Featuresd | Common (Seen With Both Acute and Chronic GVHD) |
|---|
| ALT = alanine aminotransferase; AST = aspartate aminotransferase; GI = gastrointestinal; ULN = upper limit of normal. |
| a–e See definitions in Table 6. |
| Mouth | Lichen-type features | Xerostomia | | Gingivitis |
| Hyperkeratotic plaques | Mucocele | Mucositis |
| Restriction of mouth opening from sclerosis | Pseudomembranese | Erythema |
| Mucosal atrophy | Pain |
| Ulcerse |
|
| GI Tract | Esophageal web | | Exocrine pancreatic insufficiency | Anorexia |
| Strictures or stenosis in the upper to mid third of the esophaguse | Nausea |
| Vomiting |
| Diarrhea |
| Weight loss |
| Failure to thrive (infants and children) |
| Total bilirubin, alkaline phosphatase >2 × ULNe |
| ALT or AST >2 × ULNe |
Table 8. Chronic Graft-Versus-Host Disease (GVHD) Symptoms in the Eyesa| Organ or Site | Diagnosticb | Distinctivec | Other Featuresd | Common (Seen With Both Acute and Chronic GVHD) |
|---|
| a–f See definitions in Table 6. |
| Eyes | | New onset dry, gritty, or painful eyesf | Blepharitis (erythema of the eyelids with edema) | |
| Cicatricial conjunctivitis |
| Keratoconjunctivitis siccaf | Photophobia |
| Confluent areas of punctate keratopathy | Periorbital hyperpigmentation |
Table 9. Chronic Graft-Versus-Host Disease (GVHD) Symptoms in the Genitaliaa| Organ or Site | Diagnosticb | Distinctivec | Other Featuresd | Common (Seen With Both Acute and Chronic GVHD) |
|---|
| a–e See definitions in Table 6. |
| Genitalia | Lichen planus–like features | Erosionse | | |
| Vaginal scarring or stenosis | Fissurese |
| Ulcerse |
Table 10. Chronic Graft-Versus-Host Disease (GVHD) Symptoms in the Lung, Muscles, Fascia, Joints, Hematopoietic and Immune Systems, and Other Symptomsa| Organ or Site | Diagnosticb | Distinctivec | Other Featuresd | Common (Seen With Both Acute and Chronic GVHD) |
|---|
| AIHA = autoimmune hemolytic anemia; BOOP = bronchiolitis obliterans–organizing pneumonia; ITP = idiopathic thrombocytopenic purpura; PFTs = pulmonary function tests. |
| a–f See definitions in Table 6. |
| Lung | Bronchiolitis obliterans diagnosed with lung biopsy | Bronchiolitis obliterans diagnosed with PFTs and radiologyf | | BOOP |
|
| Muscles, fascia, joints | Fasciitis | Myositis or polymyositisf | Edema | |
| Muscle cramps |
| Arthralgia or arthritis |
|
| Hematopoietic and immune | | | Thrombocytopenia | |
| Eosinophilia |
| Lymphopenia |
| Hypo- or hypergammaglobulinemia |
| Autoantibodies (AIHA and ITP) |
|
| Other | | | Pericardial or pleural effusions | |
| Ascites |
| Peripheral neuropathy |
| Nephrotic syndrome |
| Myasthenia gravis |
| Cardiac conduction abnormality or cardiomyopathy |
Risk Factors for Chronic GVHD
Chronic GVHD occurs in approximately 15% to 30% of children after sibling-donor HSCT [5] and in 20% to 45% of children after unrelated-donor HSCT. There is a higher risk of chronic GVHD with peripheral blood stem cells (PBSCs) and a lower risk with cord blood and selected approaches to haploidentical HSCT.[6,7,8]
Risk factors for the development of chronic GVHD include the following:[5,9,10]
- Patient's age (older than 10 years).
- Type of donor (unrelated and mismatched donors).
- Use of PBSCs.
- History of acute GVHD.
- Conditioning regimen (myeloablative and total-body irradiation (TBI)–based regimens).
Several factors have been associated with increased risk of nonrelapse mortality in children who develop significant chronic GVHD. Children who received HLA-mismatched grafts, received PBSCs, were older than 10 years, or had platelet counts lower than 100,000/µL at diagnosis of chronic GVHD have an increased risk of nonrelapse mortality.
The nonrelapse mortality rates were 17% at 1 year, 22% at 3 years, and 24% at 5 years after diagnosis of chronic GVHD. Many of these children required long-term immune suppression. By 3 years after diagnosis of chronic GVHD, about a third of children had died of either relapse or nonrelapse mortality, a third were off immune suppression, and a third still required some form of immune suppressive therapy.[11]
Older literature describes chronic GVHD as either limited or extensive. A National Institutes of Health (NIH) Consensus Workshop in 2006 broadened the description of chronic GVHD to three categories to better predict long-term outcomes.[12] The three NIH grading categories are as follows:[3]
- Mild disease: Involving only one or two sites, with no significant functional impairment (maximum severity score of 1 on a scale of 0 to 3).
- Moderate disease: Either involving more sites (>2) or associated with higher severity score (maximum score of 2 in any site).
- Severe disease: Indicating major disability (a score of 3 in any site or a lung score of 2).
Thus, high-risk patients include those with severe disease of any site or extensive involvement of multiple sites, especially those with the following:
- Symptomatic lung involvement.
- Skin involvement greater than 50%.
- Platelet count lower than 100,000/µL.
- Poor performance score (<60%).
- Weight loss of more than 15%.
- Chronic diarrhea.
- Progressive-onset chronic GVHD.
- History of steroid treatment with more than 0.5 mg/kg of prednisone per day for acute GVHD.
One study demonstrated a much higher chance of long-term GVHD-free survival and lower treatment-related mortality in children with mild and moderate chronic GVHD than in children with severe chronic GVHD. At 8 years, the probability of continued chronic GVHD was 4% for children with mild chronic GVHD, 11% for children with moderate chronic GVHD, and 36% for children with severe chronic GVHD.[13] In another large prospective trial with central review that used the NIH consensus criteria, about 28% of patients were misclassified as having chronic GVHD when they actually had late-acute GVHD. Additionally, there were significant challenges when using the NIH consensus criteria for bronchiolitis obliterans in children.[14]
Treatment of Chronic GVHD
Steroids remain the cornerstone of chronic GVHD therapy. However, many approaches have been developed to minimize steroid dosing, including the use of calcineurin inhibitors.[15] Topical therapy to affected areas is preferred for patients with limited disease.[16] The following agents have been tested with some success:
- Mycophenolate mofetil.[17]
- Pentostatin.[18]
- Sirolimus.[19]
- Rituximab.[20]
- Ibrutinib.[21]
- Ruxolitinib.[22]
- Belumosudil.[23]
Other approaches, including extracorporeal photopheresis, have been evaluated and show some efficacy in some patients.[24]
A series of drugs have been approved for the treatment of chronic GVHD in children.
Evidence (treatment of chronic GVHD in children):
- Ibrutinib is indicated for pediatric patients aged 1 year or older with chronic GVHD after failure of one or more lines of systemic therapy. Efficacy of ibrutinib was evaluated in an open-label multicenter trial of pediatric and young adult patients with moderate or severe chronic GVHD. The study included 47 patients who had one or more lines of therapy fail. The median age was 13 years (range, 1–19 years).[25]
- The overall response rate was 60% (95% confidence interval [CI], 44%–74%) by week 25.
- The median duration of response was 5.3 months (95% CI, 2.8–8.8).
- The median time from first response to death or new systemic therapies for chronic GVHD was 14.8 months.
- The U.S. Food and Drug Administration (FDA) approved ruxolitinib for the treatment of chronic GVHD in adults and children older than 12 years after failure of one or two lines of systemic therapy. The approval was based on one study that randomly assigned 329 patients to receive either ruxolitinib or best available therapy.[22]
- The overall response rate was 70% (95% CI, 63%–77%) for patients in the ruxolitinib arm and 57% (95% CI, 49%–65%) for patients in the best available therapy arm.
- Belumosudil, a kinase inhibitor, was approved to treat chronic GVHD in adult and pediatric patients aged 12 years and older after failure of at least two prior lines of systemic therapy. The approval was based on a study of 65 patients with chronic GVHD that was refractory to multiple lines of therapy.[23]
- The overall response rate was 74% (95%, 62%–84%) for patients who received 200 mg of belumosudil once per day and 77% (95% CI, 65%–87%) for patients who received 200 mg of belumosudil twice per day. High response rates were observed in all subgroups of patients.
- All affected organs demonstrated complete responses.
No comparative studies have been performed with these three agents. Therefore, the best drug for specific types of chronic GVHD in children has yet to be determined.
Besides significantly affecting organ function, quality of life, and functional status, infection is the major cause of chronic GVHD–related death. Therefore, all patients with chronic GVHD receive prophylaxis against Pneumocystis jirovecii pneumonia, common encapsulated organisms, and varicella by using agents such as trimethoprim/sulfamethoxazole, penicillin, and acyclovir.
Transplant-related complications account for 70% of the deaths in patients with chronic GVHD.[5] Guidelines concerning ancillary therapy and supportive care of patients with chronic GVHD have been published.[16,26]
References:
- Shlomchik WD, Lee SJ, Couriel D, et al.: Transplantation's greatest challenges: advances in chronic graft-versus-host disease. Biol Blood Marrow Transplant 13 (1 Suppl 1): 2-10, 2007.
- Bolaños-Meade J, Vogelsang GB: Chronic graft-versus-host disease. Curr Pharm Des 14 (20): 1974-86, 2008.
- Filipovich AH, Weisdorf D, Pavletic S, et al.: National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 11 (12): 945-56, 2005.
- Shulman HM, Kleiner D, Lee SJ, et al.: Histopathologic diagnosis of chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: II. Pathology Working Group Report. Biol Blood Marrow Transplant 12 (1): 31-47, 2006.
- Zecca M, Prete A, Rondelli R, et al.: Chronic graft-versus-host disease in children: incidence, risk factors, and impact on outcome. Blood 100 (4): 1192-200, 2002.
- Eapen M, Logan BR, Confer DL, et al.: Peripheral blood grafts from unrelated donors are associated with increased acute and chronic graft-versus-host disease without improved survival. Biol Blood Marrow Transplant 13 (12): 1461-8, 2007.
- Eapen M, Rubinstein P, Zhang MJ, et al.: Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet 369 (9577): 1947-54, 2007.
- Bertaina A, Zecca M, Buldini B, et al.: Unrelated donor vs HLA-haploidentical α/β T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood 132 (24): 2594-2607, 2018.
- Leung W, Ahn H, Rose SR, et al.: A prospective cohort study of late sequelae of pediatric allogeneic hematopoietic stem cell transplantation. Medicine (Baltimore) 86 (4): 215-24, 2007.
- Arora M, Klein JP, Weisdorf DJ, et al.: Chronic GVHD risk score: a Center for International Blood and Marrow Transplant Research analysis. Blood 117 (24): 6714-20, 2011.
- Jacobsohn DA, Arora M, Klein JP, et al.: Risk factors associated with increased nonrelapse mortality and with poor overall survival in children with chronic graft-versus-host disease. Blood 118 (16): 4472-9, 2011.
- Pavletic SZ, Martin P, Lee SJ, et al.: Measuring therapeutic response in chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: IV. Response Criteria Working Group report. Biol Blood Marrow Transplant 12 (3): 252-66, 2006.
- Inagaki J, Moritake H, Nishikawa T, et al.: Long-Term Morbidity and Mortality in Children with Chronic Graft-versus-Host Disease Classified by National Institutes of Health Consensus Criteria after Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant 21 (11): 1973-80, 2015.
- Cuvelier GDE, Nemecek ER, Wahlstrom JT, et al.: Benefits and challenges with diagnosing chronic and late acute GVHD in children using the NIH consensus criteria. Blood 134 (3): 304-316, 2019.
- Koc S, Leisenring W, Flowers ME, et al.: Therapy for chronic graft-versus-host disease: a randomized trial comparing cyclosporine plus prednisone versus prednisone alone. Blood 100 (1): 48-51, 2002.
- Couriel D, Carpenter PA, Cutler C, et al.: Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 12 (4): 375-96, 2006.
- Martin PJ, Storer BE, Rowley SD, et al.: Evaluation of mycophenolate mofetil for initial treatment of chronic graft-versus-host disease. Blood 113 (21): 5074-82, 2009.
- Jacobsohn DA, Gilman AL, Rademaker A, et al.: Evaluation of pentostatin in corticosteroid-refractory chronic graft-versus-host disease in children: a Pediatric Blood and Marrow Transplant Consortium study. Blood 114 (20): 4354-60, 2009.
- Jurado M, Vallejo C, Pérez-Simón JA, et al.: Sirolimus as part of immunosuppressive therapy for refractory chronic graft-versus-host disease. Biol Blood Marrow Transplant 13 (6): 701-6, 2007.
- Cutler C, Miklos D, Kim HT, et al.: Rituximab for steroid-refractory chronic graft-versus-host disease. Blood 108 (2): 756-62, 2006.
- Miklos D, Cutler CS, Arora M, et al.: Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 130 (21): 2243-2250, 2017.
- Zeiser R, Polverelli N, Ram R, et al.: Ruxolitinib for Glucocorticoid-Refractory Chronic Graft-versus-Host Disease. N Engl J Med 385 (3): 228-238, 2021.
- Cutler C, Lee SJ, Arai S, et al.: Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: the ROCKstar Study. Blood 138 (22): 2278-2289, 2021.
- González Vicent M, Ramirez M, Sevilla J, et al.: Analysis of clinical outcome and survival in pediatric patients undergoing extracorporeal photopheresis for the treatment of steroid-refractory GVHD. J Pediatr Hematol Oncol 32 (8): 589-93, 2010.
- Carpenter PA, Kang HJ, Yoo KH, et al.: Ibrutinib Treatment of Pediatric Chronic Graft-versus-Host Disease: Primary Results from the Phase 1/2 iMAGINE Study. Transplant Cell Ther 28 (11): 771.e1-771.e10, 2022.
- Carpenter PA, Kitko CL, Elad S, et al.: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 21 (7): 1167-87, 2015.
Late Mortality After Hematopoietic Stem Cell Transplant (HSCT)
The highest incidence of mortality after HSCT occurs in the first 2 years and is mostly caused by relapse. A study of late mortality (≥2 years posttransplant) in children with malignancies who underwent HSCT showed that approximately 20% of the 479 patients who were alive at 2 years had a late death. The late mortality rate was 15% in the allogeneic HSCT group (median follow-up, 10.0 years [2.0–25.6]), mainly caused by relapse (65%). A total of 26% of patients had a late death after autologous HSCT (median follow-up, 6.7 years [2.0–22.2]),[1] and recurrence of the primary malignancy accounted for 88% of these deaths. Nonrelapse mortality, death caused by chronic graft-versus-host disease (GVHD), and secondary malignancies are less common in children.
Another study reviewed the causes of late mortality after a second allogeneic transplant.[2] Of the children who were alive and relapse free 1 year after a second HSCT, 55% remained alive at 10 years. The most common cause of mortality between 1 and 10 years after HSCT in this group was relapse (77% of deaths), generally occurring in the first 3 years after transplant. The cumulative incidence of nonrelapse mortality at 10 years was 10% for this cohort. Chronic GVHD occurred in 43% of children in this study and was the leading cause of nonrelapse mortality.
One study focused on late mortality in children after autologous HSCT. The study showed that mortality rates of children who underwent transplant remained elevated compared with those of the general population more than 10 years after the procedure. However, their mortality rates approached the rates of the general population at 15 years. The study also showed a decrease in late mortality in the more current treatment eras (before 1990, 35.1%; 1990–1999, 25.6%; 2000–2010, 21.8%; P = .05).[3]
References:
- Schechter T, Pole JD, Darmawikarta D, et al.: Late mortality after hematopoietic SCT for a childhood malignancy. Bone Marrow Transplant 48 (10): 1291-5, 2013.
- Duncan CN, Majhail NS, Brazauskas R, et al.: Long-term survival and late effects among one-year survivors of second allogeneic hematopoietic cell transplantation for relapsed acute leukemia and myelodysplastic syndromes. Biol Blood Marrow Transplant 21 (1): 151-8, 2015.
- Holmqvist AS, Chen Y, Wu J, et al.: Late mortality after autologous blood or marrow transplantation in childhood: a Blood or Marrow Transplant Survivor Study-2 report. Blood 131 (24): 2720-2729, 2018.
Late Effects After Hematopoietic Stem Cell Transplant (HSCT) in Children
Data from studies of child and adult survivors of HSCT have shown that treatment-related exposures have a significant impact on survival and quality of life.[1] In one study of patients who were alive 2 years after undergoing HSCT, survivors had a 9.9-fold increased risk of premature death compared with age- and sex-matched controls in the U.S. general population.[2] Another multicenter study showed that more than one-half of adult survivors who underwent HSCT during childhood would have a grade 3 or 4 chronic health issue. Survivors had an odds ratio (OR) of 15.1 compared with siblings.[3]
Methodological Challenges in the Study of Late Effects After HSCT
Although the main cause of death in patients who have undergone HSCT is from relapse of the primary disease, many of these patients die from infections related to graft-versus-host disease (GVHD), second malignancies, or cardiac or pulmonary issues.[2,4,5,6] In addition, other studies have revealed that up to 40% of HSCT survivors experience severe, disabling, and/or life-threatening events or die because of an adverse event associated with primary or previous cancer treatment.[7,8]
Before studies aimed at decreasing the incidence and severity of these effects are initiated, it is important to understand what leads to the development of these complications:
- Pretransplant therapy: Pretransplant therapy plays an important role, but the details of significant exposures associated with pre-HSCT therapy are not included in many studies.[9]
- Preparative regimen: The transplant preparative regimen itself, including total-body irradiation (TBI) and high-dose chemotherapy, has often been studied, but this intense therapy is only a small part of a long course of therapy filled with potential causes of late effects.
- Allogenicity: The effect of allogenicity—differences in major and minor HLA antigens that lead to GVHD, autoimmunity, chronic inflammation, and, sometimes, undetected organ damage—also contributes to these late effects.
- Extended exposure to nonchemotherapeutic agents: Patients who undergo transplants may receive immunosuppressants that have significant toxicity for an extended period of time (e.g., cyclosporine or tacrolimus, which can cause hypertension and kidney damage). In addition, it is routine for patients to receive extended courses of supportive medications or antimicrobials that can be associated with organ damage (e.g., liposomal amphotericin B). These medications should be considered when assessing the risk of late effects.
Individuals differ in their susceptibility to specific organ damage from chemotherapy or in their risk of GVHD based on genetic differences in both the donor and recipient.[9,10,11]
Cardiovascular System Late Effects
Although cardiac dysfunction has been studied extensively in non-HSCT settings, less is known about the incidence and predictors of congestive heart failure following HSCT in childhood. Potentially cardiotoxic exposures unique to HSCT include the following:[12]
- Conditioning with high-dose chemotherapy, especially cyclophosphamide.
- TBI.
HSCT survivors are at increased risk of developing cardiovascular risk factors such as hypertension and diabetes, partly as a result of exposure to TBI and prolonged immunosuppressive therapy after allogeneic HSCT or related to other health conditions (e.g., hypothyroidism or growth hormone deficiency).[8,12] In a study of 661 pediatric patients who survived at least 2 years after allogeneic HSCT, 52% of patients had obesity or were overweight at their most recent examination, 18% of patients had dyslipidemia (associated with pre-HSCT anthracycline or cranial or chest irradiation), and 7% of patients were diagnosed with diabetes.[13]
Rates of cardiovascular outcomes were examined among nearly 1,500 transplant survivors (surviving ≥2 years) who were treated in Seattle from 1985 to 2006. The survivors and a population-based comparison group were matched by age, year, and sex.[14] Survivors experienced increased rates of cardiovascular death (adjusted incidence rate difference, 3.6 per 1,000 person-years [95% confidence interval, 1.7–5.5]). Survivors also had an increased cumulative incidence of the following:
- Ischemic heart disease.
- Cardiomyopathy/heart failure.
- Stroke.
- Vascular diseases.
- Rhythm disorders.
Survivors also had an increased cumulative incidence of related conditions that increased their risk of developing more serious cardiovascular disease (i.e., hypertension, renal disease, dyslipidemia, and diabetes).[14]
In addition, cardiac function and pre-HSCT exposures to chemotherapy and radiation therapy have been shown to significantly impact post-HSCT cardiac function. In evaluating post-HSCT patients for long-term issues, it is important to consider levels of pre-HSCT anthracycline and chest irradiation.[15] Although more specific studies are needed to verify this approach, current evidence suggests that the risk of late-occurring cardiovascular complications after HSCT may largely result from pre-HSCT therapeutic exposures, with little additional risk from conditioning-related exposures or GVHD.[16,17]
For more information, see the Late Effects of the Cardiovascular System section in Late Effects of Treatment for Childhood Cancer.
Neurocognitive Late Effects
Many studies report normal neurodevelopment after HSCT, with no evidence of decline.[18,19,20,21,22,23,24,25]
Researchers from St. Jude Children's Research Hospital have reported on the largest longitudinal cohort to date, describing remarkable stability in global cognitive function and academic achievement during 5 years of posttransplant follow-up.[21,22,23] This research group reported poorer outcomes in patients who underwent unrelated-donor transplant when the patients received TBI and when they experienced GVHD. But these effects on outcomes were small compared with the much larger effects of socioeconomic status on cognitive function.[22] Most published studies report similar outcomes. Normal cognitive function and academic achievement were reported in a cohort of 47 patients monitored prospectively through 2 years post-HSCT.[25] Stable cognitive function was also noted in a large cohort monitored from pretransplant to 2 years post-HSCT.[20] A smaller study reported similar normal functioning and the absence of declines over time in HSCT survivors.[18] HSCT survivors did not differ from their siblings in cognitive and academic function, with the exception that survivors performed better than siblings on measures of perceptual organization.[19] Based on findings to date, it appears that HSCT poses low-to-minimal risk of late cognitive and academic deficits in survivors.
Several studies, however, have reported some decline in cognitive function after HSCT.[26,27,28,29,30,31,32] These studies tended to include samples with a high percentage of very young children. One study reported a significant decline in IQ in their cohort at 1 year post-HSCT, deficits that were maintained at 3 years post-HSCT.[27,28] Similarly, studies from Sweden have reported deficits in visual-spatial domains and executive functioning in very young children who underwent transplant with TBI.[30,31] Another study from St. Jude Children's Research Hospital reported that while all children younger than 3 years had a decline in IQ at 1 year after transplant, patients who did not receive TBI during conditioning recovered later. Patients who received TBI had a significantly lower IQ at 5 years (P = .05) than did those who did not receive TBI.[32]
For more information, see the Hematopoietic stem cell transplant (HSCT) section in Late Effects of Treatment for Childhood Cancer.
Digestive System Late Effects
Gastrointestinal, biliary, and pancreatic dysfunction
Most gastrointestinal late effects are related to protracted acute GVHD and chronic GVHD (see Table 11). For more information, see the Hepatobiliary section in Late Effects of Treatment for Childhood Cancer.
As GVHD is controlled and tolerance is developed, most symptoms resolve. Major hepatobiliary concerns include the consequences of viral hepatitis acquired before or during the transplant, biliary stone disease, and focal liver lesions.[33] Viral serology and polymerase chain reaction should be performed to differentiate these from GVHD presenting with hepatocellular injury.[34]
Table 11. Causes of Gastrointestinal (GI), Hepatobiliary, and Pancreatic Problems in Long-Term Transplant Survivorsa| Problem Areas | Common Causes | Less Common Causes |
|---|
| ALT = alanine transaminase; AP = alkaline phosphatase; CMV = cytomegalovirus; GGT = gamma glutamyl transpeptidase; GVHD = graft-versus-host disease; HSV = herpes simplex virus; Mg++ = magnesium; VZV = varicella zoster virus. |
| a Reprinted fromBiology of Blood and Marrow Transplantation, Volume 17 (Issue 11), Michael L. Nieder, George B. McDonald, Aiko Kida, Sangeeta Hingorani, Saro H. Armenian, Kenneth R. Cooke, Michael A. Pulsipher, K. Scott Baker, National Cancer Institute–National Heart, Lung and Blood Institute/Pediatric Blood and Marrow Transplant Consortium First International Consensus Conference on Late Effects After Pediatric Hematopoietic Cell Transplantation: Long-Term Organ Damage and Dysfunction, Pages 1573–1584, Copyright 2011, with permission from American Society for Blood and Marrow Transplantation and Elsevier.[34] |
| Esophageal symptoms: heartburn, dysphagia, painful swallowing[35,36,37,38,39,40] | Oral chronic GVHD (mucosal changes, poor dentition, xerostomia) | Chronic GVHD of the esophagus (webs, rings, submucosal fibrosis and strictures, aperistalsis) |
| Reflux of gastric fluid | Hypopharyngeal dysmotility (myasthenia gravis, cricopharyngeal incoordination) |
| Squamous > adenocarcinoma |
| Pill esophagitis |
| Infection (fungal, viral) |
|
| Upper gut symptoms: anorexia, nausea, vomiting[41,42,43,44,45] | Protracted acute GI GVHD | Secondary adrenal insufficiency |
| Activation of latent infection (CMV, HSV, VZV) | Acquisition of infection (enteric viruses, Giardia, cryptosporidia,Haemophilus pylori) |
| Medication adverse effects | Gut dysmotility |
|
| Mid gut and colonic symptoms: diarrhea and abdominal pain[46,47] | Protracted acute GI GVHD | Acquisition of infection (enteric viruses, bacteria, parasites) |
| Activation of latent CMV, VZV | Pancreatic insufficiency |
| Drugs (mycophenolate mofetil, Mg++, antibiotics) | Clostridium difficilecolitis |
| Collagen-encased bowel (GVHD) |
| Rare: inflammatory bowel disease, sprue;[47]bile salt malabsorption; disaccharide malabsorption |
|
| Liver problems[33,48,49,50,51,52,53,54,55,56,57] | Cholestatic GVHD | Hepatitic GVHD |
| Chronic viral hepatitis (B and C) | VZV or HSV hepatitis |
| Cirrhosis | Fungal abscess |
| Focal nodular hyperplasia | Nodular regenerative hyperplasia |
| Nonspecific elevation of liver enzymes in serum (AP, ALT, GGT) | Biliary obstruction |
| Drug-induced liver injury |
|
| Biliary and pancreatic problems [58,59,60,61] | Cholecystitis | Pancreatic atrophy/insufficiency |
| Common duct stones/sludge | Pancreatitis/edema, stone or sludge related |
| Gall bladder sludge (calcium bilirubinate) | Pancreatitis, tacrolimus related |
| Gallstones |
Iron overload
Iron overload occurs in almost all patients who undergo HSCT, especially if the procedure is for a condition associated with transfusion dependence before HSCT (e.g., thalassemia, bone marrow failure syndromes) or pre-HSCT treatments requiring transfusions after myelotoxic chemotherapy (e.g., acute leukemias). Inflammatory conditions such as GVHD also increase gastrointestinal iron absorption. Non-HSCT conditions leading to iron overload can lead to cardiac dysfunction, endocrine disorders (e.g., pituitary insufficiency, hypothyroidism), diabetes, neurocognitive effects, and second malignancies.[34]
The effects of iron overload on morbidity post-HSCT have not been well studied. However, reducing iron levels after HSCT for thalassemia has been shown to improve cardiac function.[62]
Data supporting iron reduction therapies (such as phlebotomy or chelation after HSCT) have not identified specific levels at which iron reduction should be performed. However, higher levels of ferritin and/or evidence of significant iron overload by liver biopsy or T2-weighted magnetic resonance imaging (MRI) [63] should be addressed by iron reduction therapy.[64]
Endocrine System Late Effects
Thyroid dysfunction
Studies show that rates of thyroid dysfunction in children after myeloablative HSCT vary, with larger series reporting an average incidence of about 30%.[65,66,67,68,69,70,71,72,73,74,75] A lower incidence in adults (on average, 15%) and a notable increase in incidence in children younger than 10 years who underwent HSCT suggest that a developing thyroid gland may be more susceptible to damage.[65,67,71,75]
Pretransplant local thyroid radiation contributes to high rates of thyroid dysfunction in patients with Hodgkin lymphoma.[65] Early studies showed very high rates of thyroid dysfunction after high single-dose fractions of TBI,[76] but traditional fractionated TBI/cyclophosphamide compared with busulfan/cyclophosphamide showed similar rates of thyroid dysfunction, suggesting a role for high-dose chemotherapy in thyroid damage.[68,69,70] Notably, one large study showed that patients treated with either TBI or busulfan had similar high rates of thyroid dysfunction, while patients treated with treosulfan or reduced-intensity, chemotherapy-based regimens had low rates of thyroid disease.[75] For more information, see the Posttransplant thyroid dysfunction section in Late Effects of Treatment for Childhood Cancer.
Higher rates of thyroid dysfunction occur with single-drug prophylaxis than with three-drug GVHD prophylaxis.[77] Increased rates of thyroid dysfunction occur after unrelated-donor HSCT than after related-donor HSCT (36% vs. 9%),[66] suggesting a role for alloimmune damage in causing thyroid dysfunction.[70,78]
Growth impairment
Growth impairment is generally multifactorial. Factors that play a role in failure to achieve expected adult height in young children who have undergone HSCT include the following:
- Diminished growth hormone level.
- Thyroid dysfunction.
- Disruption of pubertal sex hormone production.
- Steroid therapy.
- Poor nutritional status.
The incidence of growth impairment varies from 20% to 80%, depending on age, risk factors, and the definition of growth impairment used by reporting groups.[72,73,79,80,81,82] Risk factors include the following:[68,69,80,83]
- TBI.
- Cranial irradiation.
- Younger age.
- HSCT for acute lymphoblastic leukemia.
- HSCT occurring during a pubertal growth spurt.[84]
Patients younger than 10 years at the time of HSCT are at the highest risk of growth impairment, but they also respond best to growth hormone replacement therapy. Early screening and referral of patients with signs of growth impairment to endocrinology specialists can result in significant restoration of height in younger children.[82]
For more information, see the Growth hormone deficiency section in Late Effects of Treatment for Childhood Cancer.
Abnormal body composition and metabolic syndrome
After HSCT, adult survivors have a 2.3-fold higher risk of premature cardiovascular-related death compared with the general population.[85,86] The exact etiology of cardiovascular risk and subsequent death is largely unknown. However, the development of metabolic syndrome (a constellation of central obesity, insulin resistance, glucose intolerance, dyslipidemia, and hypertension), especially insulin resistance, as a consequence of HSCT has been suggested.[87,88,89]
In studies of conventionally treated leukemia survivors compared with those who underwent HSCT, transplant survivors are significantly more likely to manifest metabolic syndrome or multiple adverse cardiac risk factors, including central adiposity, hypertension, insulin resistance, and dyslipidemia.[34,90,91] The concern over time is that survivors who develop metabolic syndrome after HSCT will be at higher risk of significant cardiovascular-related events and/or premature death from cardiovascular-related causes.
For more information, see the Metabolic Syndrome section in Late Effects of Treatment for Childhood Cancer.
Sarcopenic obesity
The association of obesity with diabetes and cardiovascular disease risk in the general population is well established, but obesity as determined by body mass index (BMI) is uncommon in long-term survivors after HSCT.[91] However, despite having a normal BMI, HSCT survivors develop significantly altered body composition that results in both an increase in total percent fat mass and a reduction in lean body mass. This finding, called sarcopenic obesity, results in a loss of myocyte insulin receptors and an increase in adipocyte insulin receptors; the latter are less efficient in binding insulin and clearing glucose, ultimately contributing to insulin resistance.[92,93,94]
Preliminary data from 119 children and young adult survivors and 81 healthy sibling controls found that HSCT survivors had significantly lower weight but no differences in BMI or waist circumference when compared with siblings.[95] HSCT survivors had a significantly higher percent fat mass and lower lean body mass than did controls. HSCT survivors were significantly more insulin resistant than were controls, and they also had a higher incidence of other cardiovascular risk factors, such as elevated total cholesterol, low-density lipoprotein cholesterol, and triglycerides. These differences were found only in patients who had received TBI as part of their transplant conditioning regimen.
Musculoskeletal System Late Effects
Low bone mineral density
Few studies have addressed low bone mineral density after HSCT in children.[96,97,98,99,100,101,102] A significant portion of children experienced reduction in total-body bone mineral density or lumbar Z-scores showing osteopenia (18%–33%) or osteoporosis (6%–21%). Although general risk factors have been described (female sex, inactivity, poor nutritional status, White or Asian ethnicity, family history, TBI, craniospinal irradiation, corticosteroid therapy, GVHD, cyclosporine, and endocrine deficiencies [e.g., growth hormone deficiency, hypogonadism]), most reported populations have been too small for multivariate analysis to test the relative importance of each factor.[103,104,105,106,107,108,109,110,111,112,113]
Some studies in adults have shown improvement over time in low bone mineral density after HSCT.[101,114,115] However, this finding has yet to be shown in children.
Treatment for children has generally included a multifactorial approach, with vitamin D and calcium supplementation, minimization of corticosteroid therapy, participation in weight-bearing exercise, and resolution of other endocrine problems. The role of bisphosphonate therapy in children with this condition is unclear.
For more information, see the Osteoporosis and Fractures section in Late Effects of Treatment for Childhood Cancer.
Osteonecrosis
Reported incidence of osteonecrosis in children after HSCT has been 1% to 14%. However, these studies were retrospective and underestimated actual incidence because patients may have been asymptomatic early in the course of the disease.[116,117,118] Two prospective studies showed an incidence of 30% and 44% with routine MRI screening of possible target joints.[100,119] Osteonecrosis generally occurs within 3 years after HSCT, with a median onset of about 1 year. The most common locations include knees (30%–40%), hips (19%–24%), and shoulders (9%). Most patients experience osteonecrosis in two or more joints.[76,116,120,121]
In one prospective report, risk factors by multivariate analysis included age (markedly increased in children older than 10 years; OR, 7.4) and presence of osteonecrosis at the time of transplant. It is important to note that pre-HSCT factors such as corticosteroid exposure are very important in determining patient risk. In this study, 14 of 44 children who developed osteonecrosis had the disease before HSCT.[119] A Center for International Blood and Marrow Transplant Research (CIBMTR) retrospective nested control study of 160 cases and 478 control children suggested older age (>5 years), female sex, and the presence of chronic GVHD as risk factors for developing osteonecrosis.[122]
Treatment has generally consisted of minimization of corticosteroid therapy and surgical joint replacement. Most patients are not diagnosed until they present with symptoms. In one study of 44 patients with osteonecrosis lesions in whom routine yearly MRI was performed, 4 resolved completely and 2 had resolution in one of multiply involved joints.[119] The observation that some lesions can heal over time suggests caution in the surgical management of asymptomatic lesions.
For more information, see the Osteonecrosis section in Late Effects of Treatment for Childhood Cancer.
Reproductive System Late Effects
Pubertal development
Delayed, absent, or incomplete pubertal development commonly occurs after HSCT. Two studies showed pubertal delay or failure in 16% of female children who received cyclophosphamide alone, 72% of those who received busulfan/cyclophosphamide, and 57% of those who underwent fractionated TBI. In males, incomplete pubertal development or failure was noted in 14% of those who received cyclophosphamide alone, 48% of those who received busulfan/cyclophosphamide, and 58% of those who underwent TBI.[74,123] Boys who received more than 24 Gy of radiation to the testicles developed azoospermia and also experienced failure of testosterone production, requiring supplementation to develop secondary sexual characteristics.[124]
Fertility
Women
Pretransplant and transplant cyclophosphamide exposure is the best-studied agent affecting fertility. Postpubertal women younger than 30 years can tolerate up to 20 g/m2 of cyclophosphamide and have preserved ovarian function. Prepubertal females can tolerate as much as 25 g/m2 to 30 g/m2. Although the additional effect added by pretransplant exposures to cyclophosphamide and other agents has not been specifically calculated in studies, these exposures plus transplant-related chemotherapy and radiation therapy lead to ovarian failure in 65% to 84% of females undergoing myeloablative HSCT.[125,126,127,128] The use of cyclophosphamide, busulfan, and TBI as part of the preparative regimen are associated with worse ovarian function. Younger age at the time of HSCT is associated with a higher chance of menarche and ovulation.[129,130] For more information, see the Ovarian function after HSCT section in Late Effects of Treatment for Childhood Cancer.
Studies of pregnancy are challenging because data seldom indicate whether individuals are trying to conceive. Nonetheless, a large study of pregnancy in pediatric and adult survivors of myeloablative transplant demonstrated conception in 32 of 708 patients (4.5%).[125] Of those trying to conceive, patients exposed to cyclophosphamide alone (total dose 6.7 g/m2 with no pretransplant exposure) had the best chance of conception (56 of 103, 54%), while those receiving myeloablative busulfan/cyclophosphamide (0 of 73, 0%) or TBI (7 of 532, 1.3%) had much lower rates of conception.
Men
The ability of men to produce functional sperm decreases with exposure to higher doses and specific types of chemotherapy. Most men will become azoospermic at a cyclophosphamide dose of 300 mg/kg.[131] After HSCT, 48% to 85% of men will experience gonadal failure.[125,131,132] One study showed that men who received cyclophosphamide conceived only 24% of the time, compared with 6.5% of men who received busulfan/cyclophosphamide and 1.3% of those who underwent TBI.[125] For more information, see the Testicular function after HSCT section in Late Effects of Treatment for Childhood Cancer.
Effect of reduced-toxicity, reduced-intensity, or nonmyeloablative regimens
Based on clear evidence of dose effect and the lowered gonadotoxicity of some reduced-toxicity chemotherapy regimens, the use of reduced-intensity, reduced-toxicity, or nonmyeloablative regimens will likely lead to a higher chance of preserved fertility after HSCT. Because use of these regimens is relatively new and mostly confined to older or sicker patients, most reports have consisted of single cases. Registry reports are beginning to describe pregnancies after these procedures.[128] In addition, a single-center study compared myeloablative busulfan/cyclophosphamide with reduced-intensity fludarabine/melphalan.[133][Level of evidence C1] Spontaneous puberty occurred in 56% of girls and 89% of boys after busulfan/cyclophosphamide, whereas 90% of girls and all of the boys in the fludarabine/melphalan group entered puberty spontaneously (P = .012). Significantly more girls (61%) who received busulfan/cyclophosphamide required hormone replacement than did girls in the fludarabine/melphalan group (10.5%; P = .012). In boys, no difference was noted between the two conditioning groups in time to follicle-stimulating hormone (FSH) elevation (median, 4 years in the fludarabine/melphalan group vs. 6 years in the busulfan/cyclophosphamide group). While the two regimens have similar effects on testicular function, ovarian function seems to be better preserved in girls undergoing HSCT with reduced-intensity conditioning approaches.
Another study compared serum concentrations of antimüllerian hormone (AMH) and inhibin B in 121 children who survived more than 1 year following a single HSCT and received a treosulfan-based regimen (treosulfan; low-toxicity), a fludarabine/melphalan regimen (Flu/Mel; reduced-intensity), or a busulphan/cyclophosphamide regimen (Bu/Cy; myeloablative). Mean age at HSCT was 3.6 years; mean age at follow-up was 11.8 years. Mean length of follow-up was 9.9 years. Mean AMH standard deviation scores (SDS) were significantly higher after treosulfan (-1.047) and Flu/Mel (-1.255) than after Bu/Cy (-1.543), suggesting less ovarian reserve impairment after treosulfan and Flu/Mel than after Bu/Cy. Mean serum AMH concentration was significantly better with treosulfan (>1.0 μg/l) than with Flu/Mel or Bu/Cy. In males, mean inhibin B SDS was significantly higher after treosulfan (-0.506) than after Flu/Mel (-2.53) or some Bu/Cy (-1.23). The authors concluded that treosulfan-based regimens may confer a more favorable outlook for gonadal reserve in both sexes than Flu/Mel or Bu/Cy regimens.[134] A similar report showed better pubertal attainment and Leydig cell function in children receiving treosulfan regimens compared with busulfan and TBI-based approaches.[135]
An additional study compared gonadal function markers after myeloablative conditioning with Bu/Cy and cyclophosphamide/TBI regimens with a reduced-intensity conditioning regimen using fludarabine/melphalan/alemtuzumab.[136]
- Female patients who received reduced-intensity conditioning were less likely to develop primary ovarian insufficiency, as demonstrated by elevated FSH (P = .02) and low estradiol (P = .01) or elevated luteinizing hormone (P = .09).
- Most females in the reduced-intensity conditioning (75%) and myeloablative conditioning (93%) groups had low AMH levels, indicating low or absent ovarian reserve.
- In males, although median levels of inhibin B were higher after reduced-intensity conditioning, they were not significantly different between the two groups. Ten of 11 males who received reduced-intensity conditioning (91%) and all ten males who received myeloablative conditioning (100%) had azoospermia or oligospermia. The median time from HSCT to semen analysis was 3.7 years (range, 1.3–12.2 years).
- Many of these patients had pre-HSCT exposures to gonadotoxic drugs that were not taken into consideration in the analysis.
- Although this study was small, it provided evidence that risk of infertility after reduced-intensity conditioning regimens such as fludarabine/melphalan/alemtuzumab may be substantial.
Respiratory System Late Effects
Chronic pulmonary dysfunction
The following two forms of chronic pulmonary dysfunction are observed after HSCT:[137,138,139,140,141,142]
- Obstructive lung disease.
- Restrictive lung disease.
The incidence of both forms of lung toxicity can range from 10% to 40%, depending on donor source, the time interval after HSCT, definition applied, and presence of chronic GVHD. In both conditions, collagen deposition and the development of fibrosis in either the interstitial space (restrictive lung disease) or the peribronchiolar space (obstructive lung disease) are believed to underlie the pathology.[143]
Obstructive lung disease
The most common form of obstructive lung disease after allogeneic HSCT is bronchiolitis obliterans.[139,142,144,145] This condition is an inflammatory process resulting in bronchiolar obliteration, fibrosis, and progressive obstructive lung disease.[137]
Historically, the term bronchiolitis obliterans was used to describe chronic GVHD of the lung, and it begins 6 to 20 months after HSCT. Pulmonary function tests show obstructive lung disease with general preservation of forced vital capacity (FVC), reductions in forced expiratory volume in 1 second (FEV1), and associated decreases in the FEV1/FVC ratio with or without significant declines in the diffusion capacity of the lung for carbon monoxide (DLCO).
Risk factors for bronchiolitis obliterans include the following:[137,144]
- Lower pretransplant FEV1/FVC values.
- Concomitant pulmonary infections.
- Chronic aspiration.
- Acute and chronic GVHD.
- Older recipient age.
- Use of mismatched donors.
- High-dose (vs. reduced-intensity) conditioning.
The clinical course of bronchiolitis obliterans is variable, but patients frequently develop progressive and debilitating respiratory failure despite the initiation of enhanced immunosuppression.
Standard treatment for obstructive lung disease combines enhanced immunosuppression with supportive care, including antimicrobial prophylaxis, bronchodilator therapy, and supplemental oxygen, when indicated.[146] The potential role for tumor necrosis factor-alpha in the pathogenesis of obstructive lung disease suggests that neutralizing agents such as etanercept may have promise.[147]
Restrictive lung disease
Restrictive lung disease is defined by reductions in FVC, total lung capacity (TLC), and DLCO. In contrast to obstructive lung disease, the FEV1/FVC ratio is maintained near 100%. Restrictive lung disease is common after HSCT and has been reported in 25% to 45% of patients by day 100.[137] Importantly, declines in TLC or FVC occurring at 100 days and 1 year after HSCT are associated with an increase in nonrelapse mortality. Early reports suggested that the incidence of restrictive lung disease increases with advancing recipient age, but subsequent studies have revealed significant restrictive lung disease in children receiving HSCT.[148]
The most recognizable form of restrictive lung disease is bronchiolitis obliterans organizing pneumonia (BOOP), more recently called cryptogenic organizing pneumonia (COP). Clinical features include dry cough, shortness of breath, and fever. Radiographic findings show diffuse, peripheral, fluffy infiltrates consistent with airspace consolidation. Although reported in fewer than 10% of HSCT recipients, the development of BOOP/COP is strongly associated with previous acute and chronic GVHD.[143]
Patients with restrictive lung disease have limited responses to multiple agents such as corticosteroids, cyclosporine, tacrolimus, and azathioprine.[146] The potential role for tumor necrosis factor-alpha in the pathogenesis of restrictive lung disease suggests that neutralizing agents such as etanercept may have promise.[147]
For more information, see the Respiratory complications associated with HSCT section in Late Effects of Treatment for Childhood Cancer.
Urinary System Late Effects
Renal disease
Chronic kidney disease is frequently diagnosed after transplant. There are many clinical forms of chronic kidney disease, but the most commonly described ones include thrombotic microangiopathy, nephrotic syndrome, calcineurin inhibitor toxicity, acute kidney injury, and GVHD-related chronic kidney disease. Various risk factors associated with the development of chronic kidney disease have been described. However, recent studies suggest that acute and chronic GVHD may be a proximal cause of renal injury.[34]
In a systematic review of 9,317 adults and children from 28 cohorts who underwent HSCT, approximately 16.6% of patients (range, 3.6% to 89%) developed chronic kidney disease, defined as a decrease in estimated glomerular filtration rate of at least 24.5 mL/min/1.73 m2 within the first year after transplant.[149] The cumulative incidence of chronic kidney disease developing approximately 5 years after transplant ranged from 4.4% to 44.3%, depending on the type of transplant and stage of chronic kidney disease.[150,151] Mortality rates among patients with chronic kidney disease in this setting were higher than those in transplant recipients who retained normal renal function, even when studies controlled for comorbidities.[152]
It is important to aggressively treat hypertension in patients post-HSCT, especially in those treated with prolonged courses of calcineurin inhibitors. Whether patients with post-HSCT albuminuria and hypertension benefit from treatment with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers requires further study, but careful control of hypertension with captopril, an ACE inhibitor, did show a benefit in a small study.[153]
Quality of Life
Health-related quality of life (HRQL)
HRQL is a multidimensional construct, incorporating a subjective appraisal of one's functioning and well-being, with reference to the impact of health issues on overall quality of life.[154,155] Many studies have shown that HRQL varies according to the following:[156]
- Time after HSCT: HRQL is worse with more recent HSCT.
- Transplant type: Unrelated-donor HSCT recipients have worse HRQL than do autologous or allogeneic related-donor HSCT recipients.
- Presence or absence of HSCT-related sequelae: HRQL is worse with chronic GVHD.
Pre-HSCT factors, such as family cohesion and a child's adaptive functioning, have been shown to affect HRQL.[157] Several groups have also identified the importance of pre-HSCT parenting stress on parental ratings of children's HRQL post-HSCT.[157,158,159,160,161] A report of the trajectories of HRQL over the 12 months after HSCT noted that the poorest HRQL was seen at 3 months post-HSCT, with steady improvement thereafter. Recipients of unrelated-donor transplants had the steepest declines in HRQL from baseline to 3 months. Another study reported that compromised emotional functioning, high levels of worry, and reduced communication during the acute recovery period had a negative impact on HRQL at 1-year post-HSCT.[162] Longitudinal studies identified an association of the following additional baseline risk factors with the trajectory of HRQL after HSCT:
- Child's age: Older children had worse HRQL.[157,163,164]
- Child's sex: Females had worse HRQL.[164]
- Rater: Mothers reported lower HRQL than did fathers; parents reported lower HRQL than did children.[165,166]
- Concordance by primary language or by sex of the raters: Concordant pairs reported higher HRQL.[167]
- Parental emotional distress: Greater parental distress led to worse HRQL.[163]
- Child's race: African American children had better HRQL.[164]
A report that investigated the impact of specific HSCT complications indicated that HRQL was worse among children with severe end-organ toxicity, systemic infection, or GVHD.[158] Cross-sectional studies reported that the HRQL among pediatric HSCT survivors of 5 years or longer was reasonably good, although psychological, cognitive, or physical problems appeared to negatively influence HRQL. Female sex, causal diagnosis for HSCT (e.g., acute myelogenous leukemia patients had worse HRQL), and intensity of pre-HSCT therapy were all identified as affecting HRQL post-HSCT.[168,169] Finally, another cross-sectional study of children 5 to 10 years post-HSCT cautioned that parental concerns about the child's vulnerability may induce overprotective parenting.[161]
Functional outcomes
Physician-reported physical performance
Clinician reports of long-term disability among childhood HSCT survivors suggest that the prevalence and severity of functional loss is low, as described in the following studies:
- A study from the European Society for Blood and Marrow Transplantation used the Karnofsky performance scale to report outcomes among 647 HSCT survivors (surviving ≥5 years).[170] In this cohort, 40% of survivors were younger than 18 years when they underwent transplant; only 19% had Karnofsky scores lower than 100. Seven percent had scores lower than 80, defined as the inability to work. Similar low rates of clinician-graded poor functional outcomes were reported by two other groups.[168,171]
- Among 50 survivors of childhood allogeneic HSCT treated at the City of Hope National Medical Center and Stanford University Hospital, all had Karnofsky scores of 90 or 100.[171]
- Among 73 young adults (mean age, 26 years) treated at the Karolinska University Hospital, the median Karnofsky score at 10 years post-HSCT was 90.[168]
Self-reported physical performance
Self-reported and proxy data among survivors of childhood HSCT indicated similar low rates of functional loss in the following studies:
- One study evaluated 22 survivors of childhood allogeneic HSCT (mean age at HSCT, 11 years; mean age at questionnaire, 25 years) and reported no differences between survivors' scores and population-expected values on standardized physical performance scales.[172]
- Another study compared a group of survivors who underwent transplant for childhood leukemia (n = 142) with a group of childhood leukemia survivors treated with chemotherapy alone (n = 288).[173] There were no differences between the groups on the physical function and leisure scales using multiple standardized measures.
Other studies that have reported functional limitations include the following:
- In the Bone Marrow Transplant Survivors Study (BMTSS) that included 235 survivors of childhood HSCT, 17% reported long-term physical performance limitations, compared with 8.7% of a sibling comparison group.[174]
- A Seattle study evaluated physical function in 214 young adults (median age at questionnaire, 28.7 years; 118 males) who underwent transplant at a median age of 11.9 years. When compared with age- and sex-matched controls, the HSCT survivors in this cohort scored one-half standard deviation lower on the physical component score of the SF-36 and the physical function and role physical subscales, quality-of-life measures.[169]
- A Swedish study also identified lower self-reported physical health among 73 young adult (median age, 26 years) HSCT survivors who were a median of 10 years after transplant. HSCT survivors scored significantly below population normative values on physical functioning (90.2 for HSCT survivors vs. 95.3 for population), satisfaction with physical health (66.0 for HSCT survivors vs. 78.7 for population), and role limitation because of physical health (72.7 for HSCT survivors vs. 84.9 for population).[168]
Measured physical performance
Objective measurements of function in the pediatric HSCT patient and survivor population hint that loss of physical capacity may be a bigger problem than revealed in studies that rely on clinician or self-report data. Studies measuring cardiopulmonary fitness have observed the following:
- One study used exercise capacity with cycle ergometry in a group of 20 children and young adults before HSCT, 31 patients at 1 year post-HSCT, and 70 healthy controls.[175] The average peak oxygen consumption was 21 mL/kg/min in the pre-HSCT group, 24 mL/kg/min in the post-HSCT group, and 34 mL/kg/min in the healthy controls. Among the HSCT survivors, 62% of those with cancer diagnoses scored in the lowest fifth percentile for peak oxygen consumption, compared with healthy controls.
- Another study examined exercise capacity with a Bruce treadmill protocol in 31 survivors of pediatric HSCT. In this cohort, 25.8% of HSCT survivors had exercise capacities in the 70% to 79% of predicted category, and 41.9% had exercise capacities in the lower than 70% of predicted category.[176]
- A third study investigated exercise capacity among 33 HSCT survivors who underwent transplant at a mean age of 11.3 years. At 5 years post-HSCT, only 4 of 33 survivors scored above the 75th percentile on a serial cycle ergometry test.[177]
Predictors of poor physical performance
The BMTSS found associations between poor physical performance outcomes and chronic GVHD, cardiac conditions, immune suppression, or treatment for a second malignant neoplasm.[178] In a study from the Fred Hutchison Cancer Research Center, poor performance was associated with myeloid disease.[169]
Published Guidelines for Long-Term Follow-Up
Several organizations have published consensus guidelines for follow-up for late effects after HSCT. The CIBMTR, along with the American Society of Blood and Marrow Transplant (ASBMT), and in cooperation with five other international transplant groups, published consensus recommendations for screening and preventive practices for long-term survivors of HSCT.[179]
Although some pediatric-specific challenges are addressed in these guidelines, many important pediatric issues are not. Some of these issues have been partially covered by general guidelines published by the Children's Oncology Group (COG) and other children's cancer groups (United Kingdom, Scotland). The COG has also published more specific recommendations for late effects surveillance after HSCT.[180] To address the lack of detailed, pediatric-specific, late-effects data and guidelines for long-term follow-up after HSCT, the Pediatric Transplantation and Cellular Therapy Consortium (PTCTC) published six detailed papers outlining existing data and summarizing recommendations from key groups (CIBMTR/ASBMT, COG, and the United Kingdom), along with expert recommendations for pediatric-specific issues.[9,34,64,181,182,183]
Although international efforts at further standardization and harmonization of pediatric-specific follow-up guidelines are under way, the PTCTC summary and guideline recommendations provide a consensus outline for monitoring children for late effects after HSCT.[64]
References:
- Sun CL, Francisco L, Kawashima T, et al.: Prevalence and predictors of chronic health conditions after hematopoietic cell transplantation: a report from the Bone Marrow Transplant Survivor Study. Blood 116 (17): 3129-39; quiz 3377, 2010.
- Bhatia S, Francisco L, Carter A, et al.: Late mortality after allogeneic hematopoietic cell transplantation and functional status of long-term survivors: report from the Bone Marrow Transplant Survivor Study. Blood 110 (10): 3784-92, 2007.
- Holmqvist AS, Chen Y, Hageman L, et al.: Severe, life-threatening, and fatal chronic health conditions after allogeneic blood or marrow transplantation in childhood. Cancer 129 (4): 624-633, 2023.
- Bhatia S, Robison LL, Francisco L, et al.: Late mortality in survivors of autologous hematopoietic-cell transplantation: report from the Bone Marrow Transplant Survivor Study. Blood 105 (11): 4215-22, 2005.
- Gooley TA, Chien JW, Pergam SA, et al.: Reduced mortality after allogeneic hematopoietic-cell transplantation. N Engl J Med 363 (22): 2091-101, 2010.
- Martin PJ, Counts GW, Appelbaum FR, et al.: Life expectancy in patients surviving more than 5 years after hematopoietic cell transplantation. J Clin Oncol 28 (6): 1011-6, 2010.
- Geenen MM, Cardous-Ubbink MC, Kremer LC, et al.: Medical assessment of adverse health outcomes in long-term survivors of childhood cancer. JAMA 297 (24): 2705-15, 2007.
- Oeffinger KC, Mertens AC, Sklar CA, et al.: Chronic health conditions in adult survivors of childhood cancer. N Engl J Med 355 (15): 1572-82, 2006.
- Bhatia S, Davies SM, Scott Baker K, et al.: NCI, NHLBI first international consensus conference on late effects after pediatric hematopoietic cell transplantation: etiology and pathogenesis of late effects after HCT performed in childhood--methodologic challenges. Biol Blood Marrow Transplant 17 (10): 1428-35, 2011.
- Behar E, Chao NJ, Hiraki DD, et al.: Polymorphism of adhesion molecule CD31 and its role in acute graft-versus-host disease. N Engl J Med 334 (5): 286-91, 1996.
- Goodman RS, Ewing J, Evans PC, et al.: Donor CD31 genotype and its association with acute graft-versus-host disease in HLA identical sibling stem cell transplantation. Bone Marrow Transplant 36 (2): 151-6, 2005.
- van Dalen EC, van der Pal HJ, Kok WE, et al.: Clinical heart failure in a cohort of children treated with anthracyclines: a long-term follow-up study. Eur J Cancer 42 (18): 3191-8, 2006.
- Duncan CN, Brazauskas R, Huang J, et al.: Late cardiovascular morbidity and mortality following pediatric allogeneic hematopoietic cell transplantation. Bone Marrow Transplant 53 (10): 1278-1287, 2018.
- Chow EJ, Mueller BA, Baker KS, et al.: Cardiovascular hospitalizations and mortality among recipients of hematopoietic stem cell transplantation. Ann Intern Med 155 (1): 21-32, 2011.
- Armenian SH, Sun CL, Francisco L, et al.: Late congestive heart failure after hematopoietic cell transplantation. J Clin Oncol 26 (34): 5537-43, 2008.
- Armenian SH, Sun CL, Mills G, et al.: Predictors of late cardiovascular complications in survivors of hematopoietic cell transplantation. Biol Blood Marrow Transplant 16 (8): 1138-44, 2010.
- Armenian SH, Sun CL, Kawashima T, et al.: Long-term health-related outcomes in survivors of childhood cancer treated with HSCT versus conventional therapy: a report from the Bone Marrow Transplant Survivor Study (BMTSS) and Childhood Cancer Survivor Study (CCSS). Blood 118 (5): 1413-20, 2011.
- Arvidson J, Kihlgren M, Hall C, et al.: Neuropsychological functioning after treatment for hematological malignancies in childhood, including autologous bone marrow transplantation. Pediatr Hematol Oncol 16 (1): 9-21, 1999 Jan-Feb.
- Barrera M, Atenafu E: Cognitive, educational, psychosocial adjustment and quality of life of children who survive hematopoietic SCT and their siblings. Bone Marrow Transplant 42 (1): 15-21, 2008.
- Kupst MJ, Penati B, Debban B, et al.: Cognitive and psychosocial functioning of pediatric hematopoietic stem cell transplant patients: a prospective longitudinal study. Bone Marrow Transplant 30 (9): 609-17, 2002.
- Phipps S, Brenner M, Heslop H, et al.: Psychological effects of bone marrow transplantation on children and adolescents: preliminary report of a longitudinal study. Bone Marrow Transplant 15 (6): 829-35, 1995.
- Phipps S, Rai SN, Leung WH, et al.: Cognitive and academic consequences of stem-cell transplantation in children. J Clin Oncol 26 (12): 2027-33, 2008.
- Phipps S, Dunavant M, Srivastava DK, et al.: Cognitive and academic functioning in survivors of pediatric bone marrow transplantation. J Clin Oncol 18 (5): 1004-11, 2000.
- Cognitive functioning after BMT. In: Pot-Mees CC: The Psychological Effects of Bone Marrow Transplantation in Children. Eburon Delft, 1989, pp 96–103.
- Simms S, Kazak AE, Golomb V, et al.: Cognitive, behavioral, and social outcome in survivors of childhood stem cell transplantation. J Pediatr Hematol Oncol 24 (2): 115-9, 2002.
- Cool VA: Long-term neuropsychological risks in pediatric bone marrow transplant: what do we know? Bone Marrow Transplant 18 (Suppl 3): S45-9, 1996.
- Kramer JH, Crittenden MR, DeSantes K, et al.: Cognitive and adaptive behavior 1 and 3 years following bone marrow transplantation. Bone Marrow Transplant 19 (6): 607-13, 1997.
- Kramer JH, Crittenden MR, Halberg FE, et al.: A prospective study of cognitive functioning following low-dose cranial radiation for bone marrow transplantation. Pediatrics 90 (3): 447-50, 1992.
- Shah AJ, Epport K, Azen C, et al.: Progressive declines in neurocognitive function among survivors of hematopoietic stem cell transplantation for pediatric hematologic malignancies. J Pediatr Hematol Oncol 30 (6): 411-8, 2008.
- Smedler AC, Bolme P: Neuropsychological deficits in very young bone marrow transplant recipients. Acta Paediatr 84 (4): 429-33, 1995.
- Smedler AC, Winiarski J: Neuropsychological outcome in very young hematopoietic SCT recipients in relation to pretransplant conditioning. Bone Marrow Transplant 42 (8): 515-22, 2008.
- Willard VW, Leung W, Huang Q, et al.: Cognitive outcome after pediatric stem-cell transplantation: impact of age and total-body irradiation. J Clin Oncol 32 (35): 3982-8, 2014.
- McDonald GB: Hepatobiliary complications of hematopoietic cell transplantation, 40 years on. Hepatology 51 (4): 1450-60, 2010.
- Nieder ML, McDonald GB, Kida A, et al.: National Cancer Institute-National Heart, Lung and Blood Institute/pediatric Blood and Marrow Transplant Consortium First International Consensus Conference on late effects after pediatric hematopoietic cell transplantation: long-term organ damage and dysfunction. Biol Blood Marrow Transplant 17 (11): 1573-84, 2011.
- Mackey JR, Desai S, Larratt L, et al.: Myasthenia gravis in association with allogeneic bone marrow transplantation: clinical observations, therapeutic implications and review of literature. Bone Marrow Transplant 19 (9): 939-42, 1997.
- Shimada K, Yokozawa T, Atsuta Y, et al.: Solid tumors after hematopoietic stem cell transplantation in Japan: incidence, risk factors and prognosis. Bone Marrow Transplant 36 (2): 115-21, 2005.
- McDonald GB, Sullivan KM, Schuffler MD, et al.: Esophageal abnormalities in chronic graft-versus-host disease in humans. Gastroenterology 80 (5 pt 1): 914-21, 1981.
- McDonald GB, Sullivan KM, Plumley TF: Radiographic features of esophageal involvement in chronic graft-vs.-host disease. AJR Am J Roentgenol 142 (3): 501-6, 1984.
- Schima W, Pokieser P, Forstinger C, et al.: Videofluoroscopy of the pharynx and esophagus in chronic graft-versus-host disease. Abdom Imaging 19 (3): 191-4, 1994 May-Jun.
- Minocha A, Mandanas RA, Kida M, et al.: Bullous esophagitis due to chronic graft-versus-host disease. Am J Gastroenterol 92 (3): 529-30, 1997.
- Patey-Mariaud de Serre N, Reijasse D, Verkarre V, et al.: Chronic intestinal graft-versus-host disease: clinical, histological and immunohistochemical analysis of 17 children. Bone Marrow Transplant 29 (3): 223-30, 2002.
- Akpek G, Chinratanalab W, Lee LA, et al.: Gastrointestinal involvement in chronic graft-versus-host disease: a clinicopathologic study. Biol Blood Marrow Transplant 9 (1): 46-51, 2003.
- Filipovich AH, Weisdorf D, Pavletic S, et al.: National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 11 (12): 945-56, 2005.
- McDonald GB, Bouvier M, Hockenbery DM, et al.: Oral beclomethasone dipropionate for treatment of intestinal graft-versus-host disease: a randomized, controlled trial. Gastroenterology 115 (1): 28-35, 1998.
- Hockenbery DM, Cruickshank S, Rodell TC, et al.: A randomized, placebo-controlled trial of oral beclomethasone dipropionate as a prednisone-sparing therapy for gastrointestinal graft-versus-host disease. Blood 109 (10): 4557-63, 2007.
- Iyer RV, Hahn T, Roy HN, et al.: Long-term use of oral beclomethasone dipropionate for the treatment of gastrointestinal graft-versus-host disease. Biol Blood Marrow Transplant 11 (8): 587-92, 2005.
- Borgaonkar MR, Duggan PR, Adams G: Differing clinical manifestations of celiac disease transmitted by bone marrow transplantation. Dig Dis Sci 51 (1): 210-2, 2006.
- Sullivan KM, Shulman HM, Storb R, et al.: Chronic graft-versus-host disease in 52 patients: adverse natural course and successful treatment with combination immunosuppression. Blood 57 (2): 267-76, 1981.
- Tomás JF, Pinilla I, GarcÃa-Buey ML, et al.: Long-term liver dysfunction after allogeneic bone marrow transplantation: clinical features and course in 61 patients. Bone Marrow Transplant 26 (6): 649-55, 2000.
- Strasser SI, Shulman HM, Flowers ME, et al.: Chronic graft-versus-host disease of the liver: presentation as an acute hepatitis. Hepatology 32 (6): 1265-71, 2000.
- Malik AH, Collins RH, Saboorian MH, et al.: Chronic graft-versus-host disease after hematopoietic cell transplantation presenting as an acute hepatitis. Am J Gastroenterol 96 (2): 588-90, 2001.
- Akpek G, Boitnott JK, Lee LA, et al.: Hepatitic variant of graft-versus-host disease after donor lymphocyte infusion. Blood 100 (12): 3903-7, 2002.
- Shulman HM, Sharma P, Amos D, et al.: A coded histologic study of hepatic graft-versus-host disease after human bone marrow transplantation. Hepatology 8 (3): 463-70, 1988 May-Jun.
- Strasser SI, Myerson D, Spurgeon CL, et al.: Hepatitis C virus infection and bone marrow transplantation: a cohort study with 10-year follow-up. Hepatology 29 (6): 1893-9, 1999.
- Ljungman P, Johansson N, Aschan J, et al.: Long-term effects of hepatitis C virus infection in allogeneic bone marrow transplant recipients. Blood 86 (4): 1614-8, 1995.
- Strasser SI, Sullivan KM, Myerson D, et al.: Cirrhosis of the liver in long-term marrow transplant survivors. Blood 93 (10): 3259-66, 1999.
- Peffault de Latour R, Lévy V, Asselah T, et al.: Long-term outcome of hepatitis C infection after bone marrow transplantation. Blood 103 (5): 1618-24, 2004.
- Ko CW, Murakami C, Sekijima JH, et al.: Chemical composition of gallbladder sludge in patients after marrow transplantation. Am J Gastroenterol 91 (6): 1207-10, 1996.
- Akpek G, Valladares JL, Lee L, et al.: Pancreatic insufficiency in patients with chronic graft-versus-host disease. Bone Marrow Transplant 27 (2): 163-6, 2001.
- Maringhini A, Gertz MA, DiMagno EP: Exocrine pancreatic insufficiency after allogeneic bone marrow transplantation. Int J Pancreatol 17 (3): 243-7, 1995.
- Radu B, Allez M, Gornet JM, et al.: Chronic diarrhoea after allogenic bone marrow transplantation. Gut 54 (1): 161, 174, 2005.
- Mariotti E, Angelucci E, Agostini A, et al.: Evaluation of cardiac status in iron-loaded thalassaemia patients following bone marrow transplantation: improvement in cardiac function during reduction in body iron burden. Br J Haematol 103 (4): 916-21, 1998.
- Angelucci E, Muretto P, Lucarelli G, et al.: Treatment of iron overload in the "ex-thalassemic". Report from the phlebotomy program. Ann N Y Acad Sci 850: 288-93, 1998.
- Pulsipher MA, Skinner R, McDonald GB, et al.: National Cancer Institute, National Heart, Lung and Blood Institute/Pediatric Blood and Marrow Transplantation Consortium First International Consensus Conference on late effects after pediatric hematopoietic cell transplantation: the need for pediatric-specific long-term follow-up guidelines. Biol Blood Marrow Transplant 18 (3): 334-47, 2012.
- Sanders JE, Hoffmeister PA, Woolfrey AE, et al.: Thyroid function following hematopoietic cell transplantation in children: 30 years' experience. Blood 113 (2): 306-8, 2009.
- Bailey HK, Kappy MS, Giller RH, et al.: Time-course and risk factors of hypothyroidism following allogeneic hematopoietic stem cell transplantation (HSCT) in children conditioned with fractionated total body irradiation. Pediatr Blood Cancer 51 (3): 405-9, 2008.
- Ishiguro H, Yasuda Y, Tomita Y, et al.: Long-term follow-up of thyroid function in patients who received bone marrow transplantation during childhood and adolescence. J Clin Endocrinol Metab 89 (12): 5981-6, 2004.
- Michel G, Socié G, Gebhard F, et al.: Late effects of allogeneic bone marrow transplantation for children with acute myeloblastic leukemia in first complete remission: the impact of conditioning regimen without total-body irradiation--a report from the Société Française de Greffe de Moelle. J Clin Oncol 15 (6): 2238-46, 1997.
- Afify Z, Shaw PJ, Clavano-Harding A, et al.: Growth and endocrine function in children with acute myeloid leukaemia after bone marrow transplantation using busulfan/cyclophosphamide. Bone Marrow Transplant 25 (10): 1087-92, 2000.
- Slatter MA, Gennery AR, Cheetham TD, et al.: Thyroid dysfunction after bone marrow transplantation for primary immunodeficiency without the use of total body irradiation in conditioning. Bone Marrow Transplant 33 (9): 949-53, 2004.
- Berger C, Le-Gallo B, Donadieu J, et al.: Late thyroid toxicity in 153 long-term survivors of allogeneic bone marrow transplantation for acute lymphoblastic leukaemia. Bone Marrow Transplant 35 (10): 991-5, 2005.
- Leung W, Ahn H, Rose SR, et al.: A prospective cohort study of late sequelae of pediatric allogeneic hematopoietic stem cell transplantation. Medicine (Baltimore) 86 (4): 215-24, 2007.
- Dvorak CC, Wright NB, Wong WB, et al.: Safety of hematopoietic stem cell transplantation in children less than three years of age. Pediatr Hematol Oncol 25 (8): 705-22, 2008.
- Sanders JE, Woolfrey AE, Carpenter PA, et al.: Late effects among pediatric patients followed for nearly 4 decades after transplantation for severe aplastic anemia. Blood 118 (5): 1421-8, 2011.
- Cattoni A, Molinari S, Gaiero A, et al.: Thyroid Disorders Following Hematopoietic Stem Cell Transplantation in Childhood: Impact of Conditioning Regimen on Thyroid Dysfunction, Volume Changes, and Occurrence of Nodules. Transplant Cell Ther 28 (8): 506.e1-506.e12, 2022.
- Socié G, Salooja N, Cohen A, et al.: Nonmalignant late effects after allogeneic stem cell transplantation. Blood 101 (9): 3373-85, 2003.
- Katsanis E, Shapiro RS, Robison LL, et al.: Thyroid dysfunction following bone marrow transplantation: long-term follow-up of 80 pediatric patients. Bone Marrow Transplant 5 (5): 335-40, 1990.
- Savani BN, Koklanaris EK, Le Q, et al.: Prolonged chronic graft-versus-host disease is a risk factor for thyroid failure in long-term survivors after matched sibling donor stem cell transplantation for hematologic malignancies. Biol Blood Marrow Transplant 15 (3): 377-81, 2009.
- Huma Z, Boulad F, Black P, et al.: Growth in children after bone marrow transplantation for acute leukemia. Blood 86 (2): 819-24, 1995.
- Giorgiani G, Bozzola M, Locatelli F, et al.: Role of busulfan and total body irradiation on growth of prepubertal children receiving bone marrow transplantation and results of treatment with recombinant human growth hormone. Blood 86 (2): 825-31, 1995.
- Cohen A, Rovelli A, Van-Lint MT, et al.: Final height of patients who underwent bone marrow transplantation during childhood. Arch Dis Child 74 (5): 437-40, 1996.
- Sanders JE, Guthrie KA, Hoffmeister PA, et al.: Final adult height of patients who received hematopoietic cell transplantation in childhood. Blood 105 (3): 1348-54, 2005.
- Cohen A, Rovelli A, Bakker B, et al.: Final height of patients who underwent bone marrow transplantation for hematological disorders during childhood: a study by the Working Party for Late Effects-EBMT. Blood 93 (12): 4109-15, 1999.
- Eggleston B, Patience M, Edwards S, et al.: Effect of myeloablative bone marrow transplantation on growth in children with sickle cell anaemia: results of the multicenter study of haematopoietic cell transplantation for sickle cell anaemia. Br J Haematol 136 (4): 673-6, 2007.
- Reusch JE: Current concepts in insulin resistance, type 2 diabetes mellitus, and the metabolic syndrome. Am J Cardiol 90 (5A): 19G-26G, 2002.
- Trevisan M, Liu J, Bahsas FB, et al.: Syndrome X and mortality: a population-based study. Risk Factor and Life Expectancy Research Group. Am J Epidemiol 148 (10): 958-66, 1998.
- Lakka HM, Laaksonen DE, Lakka TA, et al.: The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA 288 (21): 2709-16, 2002.
- Taskinen M, Saarinen-Pihkala UM, Hovi L, et al.: Impaired glucose tolerance and dyslipidaemia as late effects after bone-marrow transplantation in childhood. Lancet 356 (9234): 993-7, 2000.
- Baker KS, Ness KK, Steinberger J, et al.: Diabetes, hypertension, and cardiovascular events in survivors of hematopoietic cell transplantation: a report from the bone marrow transplantation survivor study. Blood 109 (4): 1765-72, 2007.
- Eaton SB, Cordain L, Sparling PB: Evolution, body composition, insulin receptor competition, and insulin resistance. Prev Med 49 (4): 283-5, 2009.
- Boirie Y: Physiopathological mechanism of sarcopenia. J Nutr Health Aging 13 (8): 717-23, 2009.
- Baker KS, Chow E, Goodman P, et al.: Adverse Impact of hematopoietic cell transplantation (HCT) on body composition and insulin resistance (IR) is associated with increased cardiovascular risk. [Abstract] Biol Blood Marrow Transplant 17 (2 Suppl): A-61, S174, 2011.
- Cook S, Weitzman M, Auinger P, et al.: Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988-1994. Arch Pediatr Adolesc Med 157 (8): 821-7, 2003.
- Genuth S, Alberti KG, Bennett P, et al.: Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care 26 (11): 3160-7, 2003.
- Grundy SM, Brewer HB, Cleeman JI, et al.: Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 109 (3): 433-8, 2004.
- Petryk A, Bergemann TL, Polga KM, et al.: Prospective study of changes in bone mineral density and turnover in children after hematopoietic cell transplantation. J Clin Endocrinol Metab 91 (3): 899-905, 2006.
- Bhatia S, Ramsay NK, Weisdorf D, et al.: Bone mineral density in patients undergoing bone marrow transplantation for myeloid malignancies. Bone Marrow Transplant 22 (1): 87-90, 1998.
- Nysom K, Holm K, Michaelsen KF, et al.: Bone mass after allogeneic BMT for childhood leukaemia or lymphoma. Bone Marrow Transplant 25 (2): 191-6, 2000.
- Daniels MW, Wilson DM, Paguntalan HG, et al.: Bone mineral density in pediatric transplant recipients. Transplantation 76 (4): 673-8, 2003.
- Kaste SC, Shidler TJ, Tong X, et al.: Bone mineral density and osteonecrosis in survivors of childhood allogeneic bone marrow transplantation. Bone Marrow Transplant 33 (4): 435-41, 2004.
- Perkins JL, Kunin-Batson AS, Youngren NM, et al.: Long-term follow-up of children who underwent hematopoeitic cell transplant (HCT) for AML or ALL at less than 3 years of age. Pediatr Blood Cancer 49 (7): 958-63, 2007.
- Carpenter PA, Hoffmeister P, Chesnut CH, et al.: Bisphosphonate therapy for reduced bone mineral density in children with chronic graft-versus-host disease. Biol Blood Marrow Transplant 13 (6): 683-90, 2007.
- Weilbaecher KN: Mechanisms of osteoporosis after hematopoietic cell transplantation. Biol Blood Marrow Transplant 6 (2A): 165-74, 2000.
- Rodgers C, Monroe R: Osteopenia and osteoporosis in pediatric patients after stem cell transplant. J Pediatr Oncol Nurs 24 (4): 184-9, 2007 Jul-Aug.
- McClune BL, Polgreen LE, Burmeister LA, et al.: Screening, prevention and management of osteoporosis and bone loss in adult and pediatric hematopoietic cell transplant recipients. Bone Marrow Transplant 46 (1): 1-9, 2011.
- Schulte CM, Beelen DW: Bone loss following hematopoietic stem cell transplantation: a long-term follow-up. Blood 103 (10): 3635-43, 2004.
- Schimmer AD, Minden MD, Keating A: Osteoporosis after blood and marrow transplantation: clinical aspects. Biol Blood Marrow Transplant 6 (2A): 175-81, 2000.
- Banfi A, Podestà M, Fazzuoli L, et al.: High-dose chemotherapy shows a dose-dependent toxicity to bone marrow osteoprogenitors: a mechanism for post-bone marrow transplantation osteopenia. Cancer 92 (9): 2419-28, 2001.
- Castañeda S, Carmona L, Carvajal I, et al.: Reduction of bone mass in women after bone marrow transplantation. Calcif Tissue Int 60 (4): 343-7, 1997.
- Lee WY, Baek KH, Rhee EJ, et al.: Impact of circulating bone-resorbing cytokines on the subsequent bone loss following bone marrow transplantation. Bone Marrow Transplant 34 (1): 89-94, 2004.
- Baker KS, Gurney JG, Ness KK, et al.: Late effects in survivors of chronic myeloid leukemia treated with hematopoietic cell transplantation: results from the Bone Marrow Transplant Survivor Study. Blood 104 (6): 1898-906, 2004.
- Stern JM, Sullivan KM, Ott SM, et al.: Bone density loss after allogeneic hematopoietic stem cell transplantation: a prospective study. Biol Blood Marrow Transplant 7 (5): 257-64, 2001.
- Ebeling PR, Thomas DM, Erbas B, et al.: Mechanisms of bone loss following allogeneic and autologous hemopoietic stem cell transplantation. J Bone Miner Res 14 (3): 342-50, 1999.
- Tauchmanovà L, De Rosa G, Serio B, et al.: Avascular necrosis in long-term survivors after allogeneic or autologous stem cell transplantation: a single center experience and a review. Cancer 97 (10): 2453-61, 2003.
- Kananen K, Volin L, Tähtelä R, et al.: Recovery of bone mass and normalization of bone turnover in long-term survivors of allogeneic bone marrow transplantation. Bone Marrow Transplant 29 (1): 33-9, 2002.
- Socié G, Cahn JY, Carmelo J, et al.: Avascular necrosis of bone after allogeneic bone marrow transplantation: analysis of risk factors for 4388 patients by the Société Française de Greffe de Moëlle (SFGM). Br J Haematol 97 (4): 865-70, 1997.
- Faraci M, Calevo MG, Lanino E, et al.: Osteonecrosis after allogeneic stem cell transplantation in childhood. A case-control study in Italy. Haematologica 91 (8): 1096-9, 2006.
- Enright H, Haake R, Weisdorf D: Avascular necrosis of bone: a common serious complication of allogeneic bone marrow transplantation. Am J Med 89 (6): 733-8, 1990.
- Sharma S, Yang S, Rochester R, et al.: Prevalence of osteonecrosis and associated risk factors in children before allogeneic BMT. Bone Marrow Transplant 46 (6): 813-9, 2011.
- Bürger B, Beier R, Zimmermann M, et al.: Osteonecrosis: a treatment related toxicity in childhood acute lymphoblastic leukemia (ALL)--experiences from trial ALL-BFM 95. Pediatr Blood Cancer 44 (3): 220-5, 2005.
- Mattano LA, Sather HN, Trigg ME, et al.: Osteonecrosis as a complication of treating acute lymphoblastic leukemia in children: a report from the Children's Cancer Group. J Clin Oncol 18 (18): 3262-72, 2000.
- Li X, Brazauskas R, Wang Z, et al.: Avascular necrosis of bone after allogeneic hematopoietic cell transplantation in children and adolescents. Biol Blood Marrow Transplant 20 (4): 587-92, 2014.
- Baker KS, Petryk A: Growth and development after hematopoietic cell transplantation. In: Appelbaum FR, Forman SJ, Negrin RS, et al., eds.: Thomas' Hematopoietic Cell Transplantation: Stem Cell Transplantation. 5th ed. John Wiley & Sons Inc., 2015, pp 1257-65.
- Sanders JE, Flournoy N, Thomas ED, et al.: Marrow transplant experience in children with acute lymphoblastic leukemia: an analysis of factors associated with survival, relapse, and graft-versus-host disease. Med Pediatr Oncol 13 (4): 165-72, 1985.
- Sanders JE, Hawley J, Levy W, et al.: Pregnancies following high-dose cyclophosphamide with or without high-dose busulfan or total-body irradiation and bone marrow transplantation. Blood 87 (7): 3045-52, 1996.
- Carter A, Robison LL, Francisco L, et al.: Prevalence of conception and pregnancy outcomes after hematopoietic cell transplantation: report from the Bone Marrow Transplant Survivor Study. Bone Marrow Transplant 37 (11): 1023-9, 2006.
- Salooja N, Szydlo RM, Socie G, et al.: Pregnancy outcomes after peripheral blood or bone marrow transplantation: a retrospective survey. Lancet 358 (9278): 271-6, 2001.
- Loren AW, Chow E, Jacobsohn DA, et al.: Pregnancy after hematopoietic cell transplantation: a report from the late effects working committee of the Center for International Blood and Marrow Transplant Research (CIBMTR). Biol Blood Marrow Transplant 17 (2): 157-66, 2011.
- Shalet SM, Didi M, Ogilvy-Stuart AL, et al.: Growth and endocrine function after bone marrow transplantation. Clin Endocrinol (Oxf) 42 (4): 333-9, 1995.
- Schubert MA, Sullivan KM, Schubert MM, et al.: Gynecological abnormalities following allogeneic bone marrow transplantation. Bone Marrow Transplant 5 (6): 425-30, 1990.
- Howell SJ, Shalet SM: Spermatogenesis after cancer treatment: damage and recovery. J Natl Cancer Inst Monogr (34): 12-7, 2005.
- Anserini P, Chiodi S, Spinelli S, et al.: Semen analysis following allogeneic bone marrow transplantation. Additional data for evidence-based counselling. Bone Marrow Transplant 30 (7): 447-51, 2002.
- Panasiuk A, Nussey S, Veys P, et al.: Gonadal function and fertility after stem cell transplantation in childhood: comparison of a reduced intensity conditioning regimen containing melphalan with a myeloablative regimen containing busulfan. Br J Haematol 170 (5): 719-26, 2015.
- Leiper A, Houwing M, Davies EG, et al.: Anti-Müllerian hormone and Inhibin B after stem cell transplant in childhood: a comparison of myeloablative, reduced intensity and treosulfan-based chemotherapy regimens. Bone Marrow Transplant 55 (10): 1985-1995, 2020.
- Cattoni A, Nicolosi ML, Capitoli G, et al.: Pubertal attainment and Leydig cell function following pediatric hematopoietic stem cell transplantation: a three-decade longitudinal assessment. Front Endocrinol (Lausanne) 14: 1292683, 2023.
- Bender JD, Oquendo-Del Toro H, Benoit J, et al.: Reduced-Intensity Conditioning Mitigates Risk for Primary Ovarian Insufficiency but Does Not Decrease Risk for Infertility in Pediatric and Young Adult Survivors of Hematopoietic Stem Cell Transplantation. Transplant Cell Ther 29 (2): 130.e1-130.e8, 2023.
- Yoshihara S, Yanik G, Cooke KR, et al.: Bronchiolitis obliterans syndrome (BOS), bronchiolitis obliterans organizing pneumonia (BOOP), and other late-onset noninfectious pulmonary complications following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 13 (7): 749-59, 2007.
- Chien JW, Duncan S, Williams KM, et al.: Bronchiolitis obliterans syndrome after allogeneic hematopoietic stem cell transplantation-an increasingly recognized manifestation of chronic graft-versus-host disease. Biol Blood Marrow Transplant 16 (1 Suppl): S106-14, 2010.
- Hildebrandt GC, Fazekas T, Lawitschka A, et al.: Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 46 (10): 1283-95, 2011.
- Chien JW, Martin PJ, Gooley TA, et al.: Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 168 (2): 208-14, 2003.
- Cerveri I, Zoia MC, Fulgoni P, et al.: Late pulmonary sequelae after childhood bone marrow transplantation. Thorax 54 (2): 131-5, 1999.
- Uhlving HH, Bang CL, Christensen IJ, et al.: Lung function after allogeneic hematopoietic stem cell transplantation in children: a longitudinal study in a population-based cohort. Biol Blood Marrow Transplant 19 (9): 1348-54, 2013.
- Freudenberger TD, Madtes DK, Curtis JR, et al.: Association between acute and chronic graft-versus-host disease and bronchiolitis obliterans organizing pneumonia in recipients of hematopoietic stem cell transplants. Blood 102 (10): 3822-8, 2003.
- Chien JW, Zhao LP, Hansen JA, et al.: Genetic variation in bactericidal/permeability-increasing protein influences the risk of developing rapid airflow decline after hematopoietic cell transplantation. Blood 107 (5): 2200-7, 2006.
- Hildebrandt GC, Granell M, Urbano-Ispizua A, et al.: Recipient NOD2/CARD15 variants: a novel independent risk factor for the development of bronchiolitis obliterans after allogeneic stem cell transplantation. Biol Blood Marrow Transplant 14 (1): 67-74, 2008.
- Cooke KR, Yanik G: Lung injury following hematopoietic stem cell transplantation. In: Appelbaum FR, Forman SJ, Negrin RS, et al., eds.: Thomas' Hematopoietic Cell Transplantation: Stem Cell Transplantation. 5th ed. John Wiley & Sons Inc., 2015, pp 1156-69.
- Yanik GA, Mineishi S, Levine JE, et al.: Soluble tumor necrosis factor receptor: enbrel (etanercept) for subacute pulmonary dysfunction following allogeneic stem cell transplantation. Biol Blood Marrow Transplant 18 (7): 1044-54, 2012.
- Norman BC, Jacobsohn DA, Williams KM, et al.: Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 46 (10): 1369-73, 2011.
- Ellis MJ, Parikh CR, Inrig JK, et al.: Chronic kidney disease after hematopoietic cell transplantation: a systematic review. Am J Transplant 8 (11): 2378-90, 2008.
- Choi M, Sun CL, Kurian S, et al.: Incidence and predictors of delayed chronic kidney disease in long-term survivors of hematopoietic cell transplantation. Cancer 113 (7): 1580-7, 2008.
- Ando M, Ohashi K, Akiyama H, et al.: Chronic kidney disease in long-term survivors of myeloablative allogeneic haematopoietic cell transplantation: prevalence and risk factors. Nephrol Dial Transplant 25 (1): 278-82, 2010.
- Cohen EP, Piering WF, Kabler-Babbitt C, et al.: End-stage renal disease (ESRD)after bone marrow transplantation: poor survival compar ed to other causes of ESRD. Nephron 79 (4): 408-12, 1998.
- Cohen EP, Irving AA, Drobyski WR, et al.: Captopril to mitigate chronic renal failure after hematopoietic stem cell transplantation: a randomized controlled trial. Int J Radiat Oncol Biol Phys 70 (5): 1546-51, 2008.
- Wilson IB, Cleary PD: Linking clinical variables with health-related quality of life. A conceptual model of patient outcomes. JAMA 273 (1): 59-65, 1995.
- Eisen M, Donald CA, Ware JE, et al.: Conceptualization and Measurement of Health for Children in the Health Insurance Study. Rand Corporation, 1980.
- Parsons SK, Barlow SE, Levy SL, et al.: Health-related quality of life in pediatric bone marrow transplant survivors: according to whom? Int J Cancer Suppl 12: 46-51, 1999.
- Barrera M, Atenafu E, Hancock K: Longitudinal health-related quality of life outcomes and related factors after pediatric SCT. Bone Marrow Transplant 44 (4): 249-56, 2009.
- Parsons SK, Shih MC, Duhamel KN, et al.: Maternal perspectives on children's health-related quality of life during the first year after pediatric hematopoietic stem cell transplant. J Pediatr Psychol 31 (10): 1100-15, 2006 Nov-Dec.
- Barrera M, Boyd-Pringle LA, Sumbler K, et al.: Quality of life and behavioral adjustment after pediatric bone marrow transplantation. Bone Marrow Transplant 26 (4): 427-35, 2000.
- Jobe-Shields L, Alderfer MA, Barrera M, et al.: Parental depression and family environment predict distress in children before stem cell transplantation. J Dev Behav Pediatr 30 (2): 140-6, 2009.
- Vrijmoet-Wiersma CM, Kolk AM, Grootenhuis MA, et al.: Child and parental adaptation to pediatric stem cell transplantation. Support Care Cancer 17 (6): 707-14, 2009.
- Felder-Puig R, di Gallo A, Waldenmair M, et al.: Health-related quality of life of pediatric patients receiving allogeneic stem cell or bone marrow transplantation: results of a longitudinal, multi-center study. Bone Marrow Transplant 38 (2): 119-26, 2006.
- Parsons SK, Ratichek SJ, Rodday AM: Caring for the caregiver: eHealth interventions for parents of pediatric hematopoietic stem cell transplant recipients. [Abstract] Pediatr Blood Cancer 56 (7): 1157, 2011.
- Brice L, Weiss R, Wei Y, et al.: Health-related quality of life (HRQoL): the impact of medical and demographic variables upon pediatric recipients of hematopoietic stem cell transplantation. Pediatr Blood Cancer 57 (7): 1179-85, 2011.
- Kaplan SH, Barlow S, Spetter D: Assessing functional status and health-related quality of life among school-aged children: reliability and validity of a new self-reported measure. [Abstract] Qual Life Res 4 (5): 444-45, 1995.
- Barrera M, Atenafu E, Doyle J, et al.: Differences in mothers' and fathers' health-related quality of life after pediatric SCT: a longitudinal study. Bone Marrow Transplant 47 (6): 855-9, 2012.
- Feichtl RE, Rosenfeld B, Tallamy B, et al.: Concordance of quality of life assessments following pediatric hematopoietic stem cell transplantation. Psychooncology 19 (7): 710-7, 2010.
- Löf CM, Winiarski J, Giesecke A, et al.: Health-related quality of life in adult survivors after paediatric allo-SCT. Bone Marrow Transplant 43 (6): 461-8, 2009.
- Sanders JE, Hoffmeister PA, Storer BE, et al.: The quality of life of adult survivors of childhood hematopoietic cell transplant. Bone Marrow Transplant 45 (4): 746-54, 2010.
- Duell T, van Lint MT, Ljungman P, et al.: Health and functional status of long-term survivors of bone marrow transplantation. EBMT Working Party on Late Effects and EULEP Study Group on Late Effects. European Group for Blood and Marrow Transplantation. Ann Intern Med 126 (3): 184-92, 1997.
- Schmidt GM, Niland JC, Forman SJ, et al.: Extended follow-up in 212 long-term allogeneic bone marrow transplant survivors. Issues of quality of life. Transplantation 55 (3): 551-7, 1993.
- Helder DI, Bakker B, de Heer P, et al.: Quality of life in adults following bone marrow transplantation during childhood. Bone Marrow Transplant 33 (3): 329-36, 2004.
- Michel G, Bordigoni P, Simeoni MC, et al.: Health status and quality of life in long-term survivors of childhood leukaemia: the impact of haematopoietic stem cell transplantation. Bone Marrow Transplant 40 (9): 897-904, 2007.
- Ness KK, Bhatia S, Baker KS, et al.: Performance limitations and participation restrictions among childhood cancer survivors treated with hematopoietic stem cell transplantation: the bone marrow transplant survivor study. Arch Pediatr Adolesc Med 159 (8): 706-13, 2005.
- Larsen RL, Barber G, Heise CT, et al.: Exercise assessment of cardiac function in children and young adults before and after bone marrow transplantation. Pediatrics 89 (4 Pt 2): 722-9, 1992.
- Eames GM, Crosson J, Steinberger J, et al.: Cardiovascular function in children following bone marrow transplant: a cross-sectional study. Bone Marrow Transplant 19 (1): 61-6, 1997.
- Hogarty AN, Leahey A, Zhao H, et al.: Longitudinal evaluation of cardiopulmonary performance during exercise after bone marrow transplantation in children. J Pediatr 136 (3): 311-7, 2000.
- Fraser CJ, Bhatia S, Ness K, et al.: Impact of chronic graft-versus-host disease on the health status of hematopoietic cell transplantation survivors: a report from the Bone Marrow Transplant Survivor Study. Blood 108 (8): 2867-73, 2006.
- Rizzo JD, Wingard JR, Tichelli A, et al.: Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation: joint recommendations of the European Group for Blood and Marrow Transplantation, Center for International Blood and Marrow Transplant Research, and the American Society for Blood and Marrow Transplantation (EBMT/CIBMTR/ASBMT). Bone Marrow Transplant 37 (3): 249-61, 2006.
- Chow EJ, Anderson L, Baker KS, et al.: Late Effects Surveillance Recommendations among Survivors of Childhood Hematopoietic Cell Transplantation: A Children's Oncology Group Report. Biol Blood Marrow Transplant 22 (5): 782-95, 2016.
- Bunin N, Small T, Szabolcs P, et al.: NCI, NHLBI/PBMTC first international conference on late effects after pediatric hematopoietic cell transplantation: persistent immune deficiency in pediatric transplant survivors. Biol Blood Marrow Transplant 18 (1): 6-15, 2012.
- Dvorak CC, Gracia CR, Sanders JE, et al.: NCI, NHLBI/PBMTC first international conference on late effects after pediatric hematopoietic cell transplantation: endocrine challenges-thyroid dysfunction, growth impairment, bone health, & reproductive risks. Biol Blood Marrow Transplant 17 (12): 1725-38, 2011.
- Parsons SK, Phipps S, Sung L, et al.: NCI, NHLBI/PBMTC First International Conference on Late Effects after Pediatric Hematopoietic Cell Transplantation: health-related quality of life, functional, and neurocognitive outcomes. Biol Blood Marrow Transplant 18 (2): 162-71, 2012.
Latest Updates to This Summary (09 / 27 / 2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Hematopoietic Stem Cell Transplant (HSCT)–Related Acute Complications
Added text to state that the U.S. Food and Drug Administration approved letermovir for cytomegalovirus prophylaxis in adults. There is solid experience using letermovir in children aged 12 years and older and emerging experience in children younger than 12 years (cited Russo et al. and Galaverna et al. as references 9 and 10, respectively).
Added text to state that a newer approach to preventing graft-versus-host disease (GVHD) includes administering posttransplant cyclophosphamide on days 3 and 4 after HSCT (cited Luznik et al. as reference 76). While it does not affect stem cells in the graft, cyclophosphamide eliminates or reduces the function or proliferation of alloreactive T cells, markedly decreasing rates of both acute and chronic GVHD (cited Nunes et al. as reference 77).
Added GVHD prophylaxis approaches as a new subsection.
Added Nutritional approaches to prevent GVHD as a new subsection.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the complications, graft-versus-host disease, and late effects after hematopoietic stem cell transplant for the treatment of pediatric cancer. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Complications, Graft-Versus-Host Disease, and Late Effects After Pediatric Hematopoietic Stem Cell Transplant are:
- Thomas G. Gross, MD, PhD (National Cancer Institute)
- Michael A. Pulsipher, MD (Huntsman Cancer Institute at University of Utah)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Complications, Graft-Versus-Host Disease, and Late Effects After Pediatric Hematopoietic Stem Cell Transplant. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/childhood-cancers/hp-stem-cell-transplant/gvhd. Accessed <MM/DD/YYYY>. [PMID: 35133768]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-09-27

