Incidence
Juvenile myelomonocytic leukemia (JMML) is a rare leukemia that occurs approximately ten times less frequently than acute myeloid leukemia in children. The annual incidence is about 1 to 2 cases per 1 million people.[1] JMML is the most common myeloproliferative neoplasm observed in young children, presenting at a median age of approximately 1.8 years. It occurs more commonly in boys (male-to-female ratio, approximately 2.5:1).
References:
- Passmore SJ, Chessells JM, Kempski H, et al.: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 121 (5): 758-67, 2003.
Clinical Presentation
Common clinical features at diagnosis include the following:[1]
- Hepatosplenomegaly (97%).
- Lymphadenopathy (76%).
- Pallor (64%).
- Fever (54%).
- Skin rash (36%).
Patients may also present with an elevated white blood cell count and increased circulating monocytes.[1]
References:
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
World Health Organization Classification
The World Health Organization (WHO) classifies juvenile myelomonocytic leukemia (JMML) as a RAS pathway activation–driven myeloproliferative neoplasm (MPN) of early childhood.[1]
For information about the classification system for acute myeloid leukemia (AML), see the World Health Organization (WHO) Classification System for Childhood AML section in Childhood Acute Myeloid Leukemia Treatment.
References:
- Khoury JD, Solary E, Abla O, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36 (7): 1703-1719, 2022.
Diagnostic Criteria
In children presenting with clinical features suggestive of juvenile myelomonocytic leukemia (JMML), current criteria for a definitive diagnosis are described in Table 1.[1]
Table 1. Diagnostic Criteria for JMML According to the 5th Edition of the WHO Classification of Hematolymphoid Tumors| GM-CSF = granulocyte-macrophage colony-stimulating factor; JMML = juvenile myelomonocytic leukemia; WHO = World Health Organization. |
| a Germline variants inPTPN11,KRAS, orNRAS(which cause Noonan syndrome) may lead to JMML-like transient myeloproliferative disorder. |
| b Occasional cases have heterozygous splice-site variants. |
| c Such asRRASorRRAS2. |
| d For cases that do not meet the genetic criteria or if genetic testing is not available. These individuals must meet the following criteria in addition to the clinical, hematologic, and laboratory criteria. |
| Clinical, Hematologic, and Laboratory Criteria (All Criteria Are Required for Diagnosis) |
| 1. Peripheral blood monocyte count is ≥1 × 109 /L |
| 2. Blasts and promonocytes constitute <20% of peripheral blood and bone marrow |
| 3. Clinical evidence of organ infiltration, most commonly splenomegaly |
| 4. Absence of theBCR::ABL1fusion gene |
| 5. Absence of aKMT2Arearrangement |
| Genetic Criteria (1 Criterion is Sufficient for Diagnosis) |
| 1. A variant in a component or a regulator of the canonical RAS pathway: |
| | a) A clonal somatic variant inPTPN11,KRAS, orNRASa |
| | b) A clonal somatic or germline variant inNF1and a loss of heterozygosity or compound heterozygosity inNF1 |
| | c) A clonal somatic or germline variant inCBLand a loss of heterozygosity inCBLb |
| 2. A noncanonical clonal RAS pathway pathogenic variantc or fusions that activate genes located upstream of the RAS pathway, such asALK,PDGFRB, andROS1 |
| Other Criteria (2 or More Are Required for Diagnosis)d |
| 1. Circulating myeloid (promyelocytes, myelocytes, metamyelocytes) and erythroid precursors |
| 2. Increased hemoglobin F for age |
| 3. Thrombocytopenia with hypercellular bone marrow, often with megakaryocytic hypoplasia; dysplastic features may or may not be evident |
| 4. Myeloid progenitors are hypersensitive to GM-CSF (detected by clonogenic assays or by measuring STAT5 phosphorylation in the absence or with low dose of exogenous GM-CSF) |
References:
- Khoury JD, Solary E, Abla O, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36 (7): 1703-1719, 2022.
Pathogenesis and Risk Factors
The pathogenesis of juvenile myelomonocytic leukemia (JMML) has been closely linked to activation of the RAS oncogene pathway, along with related syndromes (see Figure 1).[1,2] In addition, distinctive RNA expression and DNA methylation patterns have been reported. These patterns are correlated with clinical factors such as age and appear to be associated with prognosis.[3,4]
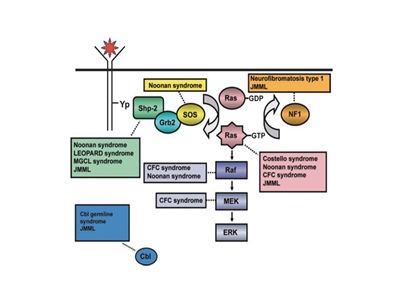
Figure 1. Schematic diagram showing ligand-stimulated Ras activation, the Ras-Erk pathway, and the gene mutations found to date contributing to the neuro-cardio-facio-cutaneous congenital disorders and JMML. NL/MGCL: Noonan-like/multiple giant cell lesion; CFC: cardia-facio-cutaneous; JMML: juvenile myelomonocytic leukemia. Reprinted from Leukemia Research, 33 (3), Rebecca J. Chan, Todd Cooper, Christian P. Kratz, Brian Weiss, Mignon L. Loh, Juvenile myelomonocytic leukemia: A report from the 2nd International JMML Symposium, Pages 355-62, Copyright 2009, with permission from Elsevier.
Syndromes and genetic features associated with an increased risk of developing JMML include the following:[5,6]
- Neurofibromatosis type 1 (NF1). Up to 14% of cases of JMML occur in children with NF1.[7]
- Noonan syndrome. Noonan syndrome is usually inherited as an autosomal dominant condition but can also arise spontaneously. It is characterized by facial dysmorphism, short stature, webbed neck, and neurocognitive and cardiac abnormalities. Germline variants in PTPN11 are observed in children with Noonan syndrome and in children with JMML.[8,9,10]
Importantly, some children with Noonan syndrome have hematologic features indistinguishable from JMML that self-resolve during infancy, similar to what happens in children with Down syndrome and transient myeloproliferative disorder.[2,10]
In a large prospective cohort of 641 patients with Noonan syndrome and a germline PTPN11 variant, 36 patients (approximately 6%) showed myeloproliferative features, with 20 patients (approximately 3%) meeting the consensus diagnostic criteria for JMML.[10]
- Of the 20 patients meeting the criteria for JMML, 12 patients had severe neonatal manifestations (e.g., life-threatening complications related to congenital heart defects, pleural effusion, leukemia infiltrates, and/or thrombocytopenia), and 10 of 20 patients died during the first month of life.
- Among the remaining eight patients, none required intensive therapy at diagnosis or during follow-up.
- All 16 patients with myeloproliferative features that did not meet JMML criteria were alive, with a median follow-up of 3 years, and no patient received chemotherapy.
- Variants in the CBL gene. CBL is an E3 ubiquitin-protein ligase that is involved in targeting proteins, particularly tyrosine kinases, for proteasomal degradation. Variants in the CBL gene occur in 10% to 15% of JMML cases,[11,12] with many of these cases occurring in children with germline CBL variants.[13,14,15]
CBL germline variants result in an autosomal dominant developmental disorder that is often characterized by impaired growth, developmental delay, cryptorchidism, and a predisposition to JMML.[13,15] Some individuals with CBL germline variants experience spontaneous regression of their JMML but develop vasculitis later in life,[13] whereas patients with only somatic CBL variants require therapy.[15] JMML arising from germline variants is clinically indistinguishable from JMML arising from somatic variants, which necessitates studies of both normal and leukemic tissue.[15]CBL variants are nearly always mutually exclusive of RAS and PTPN11 variants.[11]
References:
- Chan RJ, Cooper T, Kratz CP, et al.: Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leuk Res 33 (3): 355-62, 2009.
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.
- Bresolin S, Zecca M, Flotho C, et al.: Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol 28 (11): 1919-27, 2010.
- Olk-Batz C, Poetsch AR, Nöllke P, et al.: Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 117 (18): 4871-80, 2011.
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.
- Choong K, Freedman MH, Chitayat D, et al.: Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol 21 (6): 523-7, 1999 Nov-Dec.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Tartaglia M, Niemeyer CM, Fragale A, et al.: Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34 (2): 148-50, 2003.
- Kratz CP, Niemeyer CM, Castleberry RP, et al.: The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106 (6): 2183-5, 2005.
- Strullu M, Caye A, Lachenaud J, et al.: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 51 (10): 689-97, 2014.
- Loh ML, Sakai DS, Flotho C, et al.: Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114 (9): 1859-63, 2009.
- Muramatsu H, Makishima H, Jankowska AM, et al.: Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 115 (10): 1969-75, 2010.
- Niemeyer CM, Kang MW, Shin DH, et al.: Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42 (9): 794-800, 2010.
- Pérez B, Mechinaud F, Galambrun C, et al.: Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 47 (10): 686-91, 2010.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
Genomics of Juvenile Myelomonocytic Leukemia (JMML)
Molecular Features of JMML
The genomic landscape of JMML is characterized by variants in one of five genes of the RAS pathway: NF1, NRAS, KRAS, PTPN11, and CBL.[1,2,3] In a series of 118 consecutively diagnosed JMML cases with RAS pathway–activating variants, PTPN11 was the most commonly altered gene, accounting for 51% of cases (19% germline and 32% somatic) (see Figure 2).[1] Patients with NRAS variants accounted for 19% of cases, and patients with KRAS variants accounted for 15% of cases. NF1 variants accounted for 8% of cases, and CBL variants accounted for 11% of cases. Although variants among these five genes are generally mutually exclusive, 4% to 17% of cases have variants in two of these RAS pathway genes,[1,2,3] a finding that is associated with poorer prognosis.[1,3]
The variant rate in JMML leukemia cells is very low, but additional variants beyond those of the five RAS pathway genes described above are observed.[1,2,3] Secondary genomic alterations are observed for genes of the transcriptional repressor complex PRC2 (e.g., ASXL1 was altered in 7%–8% of cases). Some genes associated with myeloproliferative neoplasms in adults are also altered at low rates in JMML (e.g., SETBP1 was altered in 6%–9% of cases).[1,2,3,4]JAK3 variants are also observed in a small percentage (4%–12%) of JMML cases.[1,2,3,4] Cases with germline PTPN11 and germline CBL variants showed low rates of additional variants (see Figure 2).[1] The presence of variants beyond disease-defining RAS pathway variants is associated with an inferior prognosis.[1,2]
A report describing the genomic landscape of JMML found that 16 of 150 patients (11%) lacked canonical RAS pathway variants. Among these 16 patients, 3 were observed to have in-frame fusions involving receptor tyrosine kinases (DCTN1::ALK, RANBP2::ALK, and TBL1XR1::ROS1 gene fusions). These patients all had monosomy 7 and were aged 56 months or older. One patient with an ALK gene fusion was treated with crizotinib plus conventional chemotherapy and achieved a complete molecular remission and proceeded to allogeneic bone marrow transplant.[3]
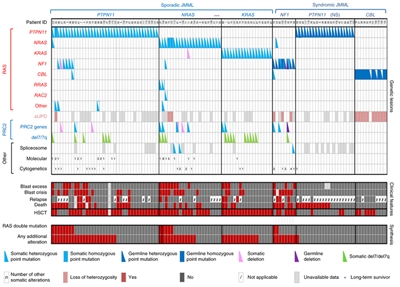
Figure 2. Alteration profiles in individual JMML cases. Germline and somatically acquired alterations with recurring hits in the RAS pathway and PRC2 network are shown for 118 patients with JMML who underwent detailed genetic analysis. Blast excess was defined as a blast count ≥10% but <20% of nucleated cells in the bone marrow at diagnosis. Blast crisis was defined as a blast count ≥20% of nucleated cells in the bone marrow. NS, Noonan syndrome. Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics (Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 [11]: 1334-40, 2015), copyright (2015).
Genomic and Molecular Prognostic Factors
Several genomic factors affect the prognosis of patients with JMML, including the following:
- Number of non–RAS pathway variants. A predictor of prognosis for children with JMML is the number of variants beyond the disease-defining RAS pathway variants.[1,2]
- One study observed that zero or one somatic alteration (pathogenic variant or monosomy 7) was identified in 64 patients (65.3%) at diagnosis, whereas two or more alterations were identified in 34 patients (34.7%).[2] In multivariate analysis, variant number (2 or more vs. 0 or 1) maintained significance as a predictor of inferior event-free survival (EFS) and overall survival (OS). A higher proportion of patients diagnosed with two or more alterations were older and male, and these patients also demonstrated a higher rate of monosomy 7 or somatic NF1 variants.[2]
- Another study observed that approximately 60% of patients had one or more additional variants beyond their disease-defining RAS pathway variant. These patients had an inferior OS compared with patients who had no additional variants (3-year OS rate, 61% vs. 85%, respectively).[1]
- A third study observed a trend for an inferior OS for patients with two or more variants compared with patients with zero or one variant.[3]
- RAS pathway double variants. Although variants in the five canonical RAS pathway genes associated with JMML (NF1, NRAS, KRAS, PTPN11, and CBL) are generally mutually exclusive, 4% to 17% of cases have variants in two of these RAS pathway genes.[1,2] This finding has been associated with a poorer prognosis.[1,2]
- Two RAS pathway variants were identified in 11% of JMML patients in one report, and these patients had a significantly inferior EFS rate (14%) compared with patients who had a single RAS pathway variant (62%). Patients with Noonan syndrome were excluded from the analyses.[2]
- Similar findings for RAS pathway variants were reported in a second study. This study observed that patients with RAS pathway double variants (15 of 96 patients) had lower survival rates than did patients with either no additional variants or with additional variants beyond the RAS pathway variant.[1]
- DNA methylation profile.
- One study applied DNA methylation profiling to a discovery cohort of 39 patients with JMML and to a validation cohort of 40 patients. Distinctive subsets of JMML with either high, intermediate, or low methylation levels were observed in both cohorts. Patients with the lowest methylation levels had the highest survival rates, and all but 1 of 15 patients experienced spontaneous resolution in the low methylation cohort. High methylation status was associated with lower EFS rates.[5]
- Another study applied DNA methylation profiling to a cohort of 106 patients with JMML. The study observed one subgroup of patients with a hypermethylation profile and one subgroup of patients with a hypomethylation profile. Patients in the hypermethylation group had a significantly lower OS rate than did patients in the hypomethylation group (5-year OS rate, 46% vs. 73%, respectively). Patients in the hypermethylation group also had a significantly poorer 5-year transplant-free survival rate than did patients in the hypomethylation group (2.2%; 95% CI, 0.2%–10.1% vs. 41.2%; 95% CI, 27.1%–54.8%). Hypermethylation status was associated with two or more variants, higher fetal hemoglobin levels, older age, and lower platelet count at diagnosis. All patients with Noonan syndrome were in the hypomethylation group.[3]
- A study examined 33 patients with JMML who had CBL variants. The study identified 31 patients with low methylation and 2 patients with intermediate methylation. Both of the children with intermediate methylation relapsed after undergoing HSCT. Because treatment, which included observation only, varied among the 31 patients with low methylation, the impact of the methylation profile on therapeutic decisions and outcomes could not be fully assessed. However, the methylation status was not prognostic of spontaneous resolution.[6]
- LIN28B overexpression. LIN28B overexpression, which is present in approximately one-half of children with JMML, identifies a biologically distinctive subset of JMML. LIN28B is an RNA-binding protein that regulates stem cell renewal.[7]
- LIN28B overexpression was positively correlated with high blood fetal hemoglobin level and age (both of which are associated with poor prognosis), and it was negatively correlated with presence of monosomy 7 (also associated with inferior prognosis). Although LIN28B overexpression identifies a subset of patients with increased risk of treatment failure, it was not found to be an independent prognostic factor when other factors such as age and monosomy 7 status are considered.[7]
- Another study also observed a subset of JMML patients with elevated LIN28B expression. The study identified LIN28B as the gene for which expression was most strongly associated with hypermethylation status.[3]
References:
- Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 (11): 1334-40, 2015.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Murakami N, Okuno Y, Yoshida K, et al.: Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131 (14): 1576-1586, 2018.
- Sakaguchi H, Okuno Y, Muramatsu H, et al.: Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45 (8): 937-41, 2013.
- Stieglitz E, Mazor T, Olshen AB, et al.: Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun 8 (1): 2127, 2017.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
- Helsmoortel HH, Bresolin S, Lammens T, et al.: LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 127 (9): 1163-72, 2016.
Clinical Prognostic Factors
Historically, more than 90% of patients with juvenile myelomonocytic leukemia (JMML) died despite the use of chemotherapy.[1] However, with the application of hematopoietic stem cell transplant, survival rates of approximately 50% are now observed.[2] Patients appeared to follow three distinct clinical courses:
- Rapidly progressive disease and early demise.
- Transiently stable disease followed by progression and death.
- Clinical improvement that lasted up to 9 years before progression or, rarely, long-term survival.
Favorable prognostic factors for survival after any therapy include the following:[3,4]
- Age younger than 2 years.
- Platelet count greater than 33 × 109 /L.
- Low age-adjusted fetal hemoglobin levels.
In contrast, being older than 2 years and having high blood fetal hemoglobin levels at diagnosis are predictors of poor outcome.[3,4]
References:
- Freedman MH, Estrov Z, Chan HS: Juvenile chronic myelogenous leukemia. Am J Pediatr Hematol Oncol 10 (3): 261-7, 1988 Fall.
- Locatelli F, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 105 (1): 410-9, 2005.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Passmore SJ, Chessells JM, Kempski H, et al.: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 121 (5): 758-67, 2003.
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has slowly increased since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence.[2] This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation to achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Pathologists.
- Pediatric radiation oncologists.
- Pediatric medical oncologists and hematologists.
- Rehabilitation specialists.
- Pediatric oncology nurses.
- Social workers.
- Child-life professionals.
- Psychologists.
- Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[3] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
References:
- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
- Wolfson J, Sun CL, Wyatt L, et al.: Adolescents and Young Adults with Acute Lymphoblastic Leukemia and Acute Myeloid Leukemia: Impact of Care at Specialized Cancer Centers on Survival Outcome. Cancer Epidemiol Biomarkers Prev 26 (3): 312-320, 2017.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed August 23, 2024.
Treatment of JMML
Treatment options for juvenile myelomonocytic leukemia (JMML) include the following:
- Chemotherapy before hematopoietic stem cell transplant (HSCT).
- HSCT.
Chemotherapy Before HSCT
Previous efforts to use chemotherapy before curative-intent HSCT have had a mixed and overall unsatisfactory impact on survival. However, control of symptoms has been aided by various lower- and higher-intensity regimens.[1,2]
Evidence (chemotherapy before HSCT):
- In an attempt to cytoreduce leukemic burden, the combination of fludarabine and cytarabine with isotretinoin alone was given for two cycles before planned HSCT in 34 of 87 evaluable children with JMML in the Children's Oncology Group (COG) AAML0122 trial.[3]
- In this group, the overall response rate (partial response [PR] and complete response [CR]) was 68%. However, achieving a CR before HSCT did not significantly improve overall survival, event-free survival (EFS), or relapse risk.
- A phase II, single-arm, open-label trial included 18 children with newly diagnosed JMML who received single-agent azacitidine, given for 7 days in 28-day cycles.[4]
- The study found that 61% of patients had partial remissions after three cycles of treatment.
- Responding patients, defined using the International JMML response criteria,[5] tended to be younger and had low-to-medium methylation classifications.
- Six patients became platelet-transfusion independent, and all responders had reductions in splenomegaly.
- Seventeen of the 18 patients received an HSCT at a median of 5.5 months after diagnosis. Of these 17 patients, 14 remained leukemia-free at last follow-up (median, 23.8 months after HSCT).[4]
Partially based on this trial, the U.S. Food and Drug Administration expanded the approved indications for azacitidine to include children with newly diagnosed JMML.
HSCT
HSCT currently offers the best chance of cure for JMML.[1,6,7,8,9]
Evidence (HSCT):
- A report from the European Working Group on Childhood Myelodysplastic Syndromes included 100 transplant recipients at multiple centers treated with a common preparative regimen of busulfan, cyclophosphamide, and melphalan, with or without antithymocyte globulin. Recipients had been treated with varying degrees of pretransplant chemotherapy or differentiating agents, and some patients had a splenectomy.[7]
- The 5-year EFS rate was 55% for children with JMML who underwent HSCT using HLA-identical matched family donor cells and 49% for children with JMML who underwent HSCT using unrelated donor cells.
- The multivariate analysis showed no effect on survival of previous acute myeloid leukemia–like chemotherapy versus low-dose chemotherapy or no chemotherapy.
- No effect on survival was observed for splenectomy pretransplant or difference in spleen size.
- No difference in outcomes was found based on related versus unrelated donors.
- Only age older than 4 years and female sex were shown to be poor prognostic factors for outcome and increased risk of relapse (relative risk [RR], 2.24 [1.07–4.69]; P = .032 for older age; RR, 2.22 [1.09–4.50]; P = .028 for females).[7]
- In one study, cord blood transplant produced the following results:[10][Level of evidence C2]
- The 5-year disease-free survival rate was 44%.
- Outcomes were improved in children younger than 1.4 years at diagnosis, those with nonmonosomy 7 karyotype, and those receiving 5/6 to 6/6 HLA-matched cord units.
- This suggests that cord blood can provide an additional donor pool for this group of children.
- The use of reduced-intensity preparative regimens to decrease the adverse side effects of transplant have also been reported in small numbers of patients, generally for patients ineligible for myeloablative HSCT.[11,12]
- The COG conducted a randomized trial in children with JMML that compared a standard-intensity preparative regimen (busulfan/cyclophosphamide/melphalan) with a reduced-intensity regimen (busulfan/fludarabine).[13]
- The trial closed to enrollment early when an interim analysis revealed a higher frequency of relapse/disease persistence (7 of 9 patients) in children who received the reduced-intensity regimen than in children who received the standard-intensity regimen (1 of 6 patients).
The role of conventional antileukemia therapy in the treatment of JMML is not defined. Determining the role of specific agents in the treatment of JMML is complicated because of the absence of consensus response criteria.[14] Some agents that have shown antileukemia activity against JMML include etoposide, cytarabine, thiopurines (thioguanine and mercaptopurine), isotretinoin, and farnesyl inhibitors, but none of these have been shown to improve outcome.[14,15,16,17,18]; [3][Level of evidence B4]
Approaches to Recurrence After HSCT or Refractory JMML
Disease recurrence is the primary cause of treatment failure for children with JMML after HSCT and occurs in 30% to 40% of cases.[6,7,8] While the role of donor lymphocyte infusions is uncertain,[19] reports indicate that approximately 50% of patients with relapsed JMML can be successfully treated with a second HSCT.[20]
In a prospective study, four children with relapsed JMML after stem cell transplant were treated with azacitidine. Three patients responded to azacitidine and were able to proceed to a second transplant.[21]
In a prospective study, ten children with relapsed or refractory JMML were treated with oral trametinib (an MEK inhibitor) daily for up to 12 28-day cycles. Five patients had objective responses (three clinical PRs and two clinical CRs) within five cycles. Two patients had stable disease. All seven patients remained alive at a median follow-up of 24 months, including three who continued to receive trametinib off study (for 6, 24, and 24 months, respectively) without proceeding to HSCT. The four patients who underwent HSCT remained in CR at a median of 24 months of follow-up. The RAS pathway variants were no longer detected in the four patients who underwent HSCT, whereas the three other patients continued to have detectable variants without progressive disease while receiving trametinib. No severe adverse events were reported.[22]
References:
- Locatelli F, Niemeyer CM: How I treat juvenile myelomonocytic leukemia. Blood 125 (7): 1083-90, 2015.
- Wintering A, Dvorak CC, Stieglitz E, et al.: Juvenile myelomonocytic leukemia in the molecular era: a clinician's guide to diagnosis, risk stratification, and treatment. Blood Adv 5 (22): 4783-4793, 2021.
- Stieglitz E, Ward AF, Gerbing RB, et al.: Phase II/III trial of a pre-transplant farnesyl transferase inhibitor in juvenile myelomonocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (4): 629-36, 2015.
- Niemeyer CM, Flotho C, Lipka DB, et al.: Response to upfront azacitidine in juvenile myelomonocytic leukemia in the AZA-JMML-001 trial. Blood Adv 5 (14): 2901-2908, 2021.
- Niemeyer CM, Loh ML, Cseh A, et al.: Criteria for evaluating response and outcome in clinical trials for children with juvenile myelomonocytic leukemia. Haematologica 100 (1): 17-22, 2015.
- Smith FO, King R, Nelson G, et al.: Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia. Br J Haematol 116 (3): 716-24, 2002.
- Locatelli F, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 105 (1): 410-9, 2005.
- Yusuf U, Frangoul HA, Gooley TA, et al.: Allogeneic bone marrow transplantation in children with myelodysplastic syndrome or juvenile myelomonocytic leukemia: the Seattle experience. Bone Marrow Transplant 33 (8): 805-14, 2004.
- Baker D, Cole C, Price J, et al.: Allogeneic bone marrow transplantation in juvenile myelomonocytic leukemia without total body irradiation. J Pediatr Hematol Oncol 26 (3): 200-3, 2004.
- Locatelli F, Crotta A, Ruggeri A, et al.: Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood 122 (12): 2135-41, 2013.
- Yabe M, Sako M, Yabe H, et al.: A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr Transplant 12 (8): 862-7, 2008.
- Koyama M, Nakano T, Takeshita Y, et al.: Successful treatment of JMML with related bone marrow transplantation after reduced-intensity conditioning. Bone Marrow Transplant 36 (5): 453-4; author reply 454, 2005.
- Dvorak CC, Satwani P, Stieglitz E, et al.: Disease burden and conditioning regimens in ASCT1221, a randomized phase II trial in children with juvenile myelomonocytic leukemia: A Children's Oncology Group study. Pediatr Blood Cancer 65 (7): e27034, 2018.
- Bergstraesser E, Hasle H, Rogge T, et al.: Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria. Pediatr Blood Cancer 49 (5): 629-33, 2007.
- Castleberry RP, Emanuel PD, Zuckerman KS, et al.: A pilot study of isotretinoin in the treatment of juvenile chronic myelogenous leukemia. N Engl J Med 331 (25): 1680-4, 1994.
- Woods WG, Barnard DR, Alonzo TA, et al.: Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol 20 (2): 434-40, 2002.
- Loh ML: Childhood myelodysplastic syndrome: focus on the approach to diagnosis and treatment of juvenile myelomonocytic leukemia. Hematology Am Soc Hematol Educ Program 2010: 357-62, 2010.
- Hasle H: Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr 19 (1): 1-8, 2007.
- Yoshimi A, Bader P, Matthes-Martin S, et al.: Donor leukocyte infusion after hematopoietic stem cell transplantation in patients with juvenile myelomonocytic leukemia. Leukemia 19 (6): 971-7, 2005.
- Yoshimi A, Mohamed M, Bierings M, et al.: Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia. Leukemia 21 (3): 556-60, 2007.
- Rubio-San-Simón A, van Eijkelenburg NKA, Hoogendijk R, et al.: Azacitidine (Vidaza®) in Pediatric Patients with Relapsed Advanced MDS and JMML: Results of a Phase I/II Study by the ITCC Consortium and the EWOG-MDS Group (Study ITCC-015). Paediatr Drugs 25 (6): 719-728, 2023.
- Stieglitz E, Lee AG, Angus SP, et al.: Efficacy of the Allosteric MEK Inhibitor Trametinib in Relapsed and Refractory Juvenile Myelomonocytic Leukemia: a Report from the Children's Oncology Group. Cancer Discov 14 (9): 1590-1598, 2024.
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT05849662 (A Phase I/II Study of Trametinib and Azacitidine for Patients With Newly Diagnosed Juvenile Myelomonocytic Leukemia [JMML]): This clinical trial will test the safety and efficacy of combining trametinib and azacitidine in patients with JMML.
Latest Updates to This Summary (12 / 10 / 2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Juvenile Myelomonocytic Leukemia (JMML)
Revised text to state that previous efforts to use chemotherapy before curative-intent hematopoietic stem cell transplant (HSCT) have had a mixed and overall unsatisfactory impact on survival. However, control of symptoms has been aided by various lower- and higher-intensity regimens (cited Wintering et al. as reference 2).
Added text to state that in an attempt to cytoreduce leukemic burden, the combination of fludarabine and cytarabine with isotretinoin alone was given for two cycles before planned HSCT in 34 of 87 evaluable children with JMML in the Children's Oncology Group AAML0122 trial. In this group, the overall response rate was 68%. However, achieving a complete response before HSCT did not significantly improve overall survival, event-free survival, or relapse risk.
Added Approaches to Recurrence After HSCT or Refractory JMML as a new subsection.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of juvenile myelomonocytic leukemia. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Juvenile Myelomonocytic Leukemia Treatment are:
- Alan Scott Gamis, MD, MPH (Children's Mercy Hospital)
- Karen J. Marcus, MD, FACR (Dana-Farber of Boston Children's Cancer Center and Blood Disorders Harvard Medical School)
- Jessica Pollard, MD (Dana-Farber/Boston Children's Cancer and Blood Disorders Center)
- Michael A. Pulsipher, MD (Huntsman Cancer Institute at University of Utah)
- Rachel E. Rau, MD (University of Washington School of Medicine, Seatle Children's)
- Lewis B. Silverman, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
- Sarah K. Tasian, MD (Children's Hospital of Philadelphia)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Juvenile Myelomonocytic Leukemia Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/leukemia/hp/child-aml-treatment-pdq/childhood-jmml-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 38630974]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-12-10

