Introduction
Birt-Hogg-Dubé syndrome (BHD) is an autosomal dominantly inherited hamartomatous disorder caused by germline pathogenic variants in the FLCNgene.[1,2] First described by Birt in 1977, BHD is characterized by cutaneous hamartomas known as fibrofolliculomas /trichodiscomas.[3] Clinical characteristics of BHD also include pulmonary cysts, spontaneous pneumothoraxes, and various histological types of renal tumors.[4]Acrochordons can be found in individuals with BHD, but they are also commonly found in the general population and are therefore, not diagnostic.[5,6,7]
Disease severity can vary significantly. Skin lesions typically appear during the third or fourth decades of life and increase in size and number with age. Lung cysts are usually bilateral and multifocal. Most individuals with lung cysts are asymptomatic but have a high risk of developing spontaneous pneumothoraxes.
Approximately 15% to 30% of individuals with BHD develop renal tumors, which are typically bilateral, multifocal, and slow growing.[8,9,10] Tumors are diagnosed at a median age of 46 to 50 years. In BHD, hybrid oncocytic renal tumors (these tumors have features of both oncocytomas and chromophobe histological cell types) (50%), chromophobe renal cell cancers (34%), and oncocytomas (9%) are diagnosed most often. Clear cell and papillary tumors have been described, but they make up less than 10% of all BHD-associated renal tumors.[8] Some families present with renal tumors and/or autosomal dominant spontaneous pneumothoraxes without cutaneous manifestations.[9,11,12]
Natural History
The clinical characteristics of BHD include fibrofolliculomas/trichodiscomas (types of cutaneous hamartomas), pulmonary cysts/histories of pneumothoraxes, and various histological types of renal tumors. BHD is characterized by phenotypic heterogeneity, and disease severity can vary significantly among family members and between families. To date, there is no evidence of increased risk of skin cancer or malignant transformation of these hamartomatous lesions.
In 2001, a family-based study showed that patients with the clinical diagnosis of BHD were seven times more likely than clinically unaffected family members to develop renal tumors.[13] Patients with clinical diagnoses of BHD were also 50 times more likely than clinically unaffected family members to develop spontaneous pneumothoraxes. This study confirmed that renal tumors and spontaneous pneumothoraxes are both major manifestations of BHD. While BHD-associated renal tumors can be aggressive, they are generally indolent. Most appropriately managed patients will require no more than one partial nephrectomy on each kidney during their lifetimes.[14] Metastatic disease, although described, is rare.[14]
References:
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Nickerson ML, Warren MB, Toro JR, et al.: Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2 (2): 157-64, 2002.
- Birt AR, Hogg GR, Dubé WJ: Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 113 (12): 1674-7, 1977.
- Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 12 (10): 558-69, 2015.
- Boza JC, Trindade EN, Peruzzo J, et al.: Skin manifestations of obesity: a comparative study. J Eur Acad Dermatol Venereol 26 (10): 1220-3, 2012.
- Sanfilippo AM, Barrio V, Kulp-Shorten C, et al.: Common pediatric and adolescent skin conditions. J Pediatr Adolesc Gynecol 16 (5): 269-83, 2003.
- Yosipovitch G, DeVore A, Dawn A: Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 56 (6): 901-16; quiz 917-20, 2007.
- Pavlovich CP, Walther MM, Eyler RA, et al.: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 26 (12): 1542-52, 2002.
- Benusiglio PR, Giraud S, Deveaux S, et al.: Renal cell tumour characteristics in patients with the Birt-Hogg-Dubé cancer susceptibility syndrome: a retrospective, multicentre study. Orphanet J Rare Dis 9: 163, 2014.
- Shuch B, Vourganti S, Ricketts CJ, et al.: Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 32 (5): 431-7, 2014.
- Graham RB, Nolasco M, Peterlin B, et al.: Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 172 (1): 39-44, 2005.
- Painter JN, Tapanainen H, Somer M, et al.: A 4-bp deletion in the Birt-Hogg-Dubé gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet 76 (3): 522-7, 2005.
- Zbar B, Alvord WG, Glenn G, et al.: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 11 (4): 393-400, 2002.
- Stamatakis L, Metwalli AR, Middelton LA, et al.: Diagnosis and management of BHD-associated kidney cancer. Fam Cancer 12 (3): 397-402, 2013.
Genetics
FLCNGene
FLCN, a novel tumor suppressor gene, comprises 14 exons located at chromosome 17p11.2.[1] In patients with Birt-Hogg-Dubé syndrome (BHD), FLCNpathogenic variants have been identified in all translated exons,[2,3,4,5] and pathogenic intronic variants have also been described.[6]FLCN encodes a 64-kDa phosphoprotein, folliculin (also known as the FLCN protein), which is highly conserved among species.
Prevalence
Fewer than 1,000 families with BHD have been described across all continents.[7,8] Studies have reported prevalence estimates between 1 in 200,000 and 1 in 500,000 for BHD. A study that analyzed data from a population-based biobank discovered that BHD's incidence may be 10 to 100 times higher than previously thought.[7] Eighty-nine percent of participants in this study did not have BHD diagnoses prior to genetic testing, which highlights the often subtle clinical manifestations associated with BHD.
Genotype-Phenotype Correlations
A correlation has not been established between specific FLCNvariants and renal, pulmonary, and cutaneous manifestations that are associated with BHD. However, individuals with a deletion in the polycytosine tract of exon 11 may have a lower risk of developing BHD-associated renal cancers than individuals with other FLCN variants.[2] However, the sample size in this study was small, and this observation was not replicated in a subsequent study from the same institution.[3] On the basis of the three major clinical manifestations (fibrofolliculomas /trichodiscomas, lung cysts/pneumothoraxes, and renal tumors), penetrance of BHD is considered to be very high. Anticipation is not known to occur in BHD.
References:
- Nickerson ML, Warren MB, Toro JR, et al.: Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2 (2): 157-64, 2002.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Maffé A, Toschi B, Circo G, et al.: Constitutional FLCN mutations in patients with suspected Birt-Hogg-Dubé syndrome ascertained for non-cutaneous manifestations. Clin Genet 79 (4): 345-54, 2011.
- Kunogi M, Kurihara M, Ikegami TS, et al.: Clinical and genetic spectrum of Birt-Hogg-Dube syndrome patients in whom pneumothorax and/or multiple lung cysts are the presenting feature. J Med Genet 47 (4): 281-7, 2010.
- Rossing M, Albrechtsen A, Skytte AB, et al.: Genetic screening of the FLCN gene identify six novel variants and a Danish founder mutation. J Hum Genet 62 (2): 151-157, 2017.
- Savatt JM, Shimelis H, Moreno-De-Luca A, et al.: Frequency of truncating FLCN variants and Birt-Hogg-Dubé-associated phenotypes in a health care system population. Genet Med 24 (9): 1857-1866, 2022.
- Muller ME, Daccord C, Taffé P, et al.: Prevalence of Birt-Hogg-Dubé Syndrome Determined Through Epidemiological Data on Spontaneous Pneumothorax and Bayes Theorem. Front Med (Lausanne) 8: 631168, 2021.
Molecular Biology
The identification of a somatic second hit in most Birt-Hogg-Dubé syndrome (BHD)-associated renal tumors strongly suggests that FLCN functions as a tumor suppressor. Both somatic single nucleotide variants in the wild-type FLCNallele and loss of heterozygosity at chromosome 17p have been identified, although the former appears to be the more common mechanism of inactivation of the second FLCN allele.[1]
The precise mechanisms by which inactivation of FLCN leads to tumorigenesis remain to be elucidated. However, folliculin, the protein product of FLCN, has been implicated as a component of the cellular energy–sensing system. Folliculin, in association with either of two novel folliculin-interacting proteins, FNIP1 and FNIP2, interacts with AMPK.[2,3] AMPK is a major cellular energy and nutrient sensor that regulates the activity of mTOR in response to these stimuli.[4] Additionally, both folliculin and FNIP1 are phosphorylated by AMPK, although the significance of this posttranslational modification is not clearly understood. The C-terminal domain of FLCN is required for its interaction with FNIP1 and FNIP2. Most, but not all, tumor-associated FLCN variants predict for a truncated protein missing this C-terminal domain or they appear to destabilize the FLCN protein.[2,5]
The effects of folliculin loss on mTOR activity have been studied by several groups. Tissue-specific activation of mTORC1 was demonstrated in a kidney-specific FLCN knockout mouse model.[6] In this model, both mTORC1 and mTORC2 were activated in renal tumors that developed in FLCN heterozygous knockout mice subsequent to loss of the wild-type allele,[7] suggesting that mTOR may play a role in the development of BHD-related tumors. A subsequent study suggested that aerobic glycolysis is upregulated as a consequence of FLCN inactivation. This glycolytic shift, although moderate, appears to be a consequence of constitutive AMPK activation in FLCN-null cells. AMPK activation has been shown to upregulate hypoxia-inducible factor 1 (HIF1) and is well studied as a transcriptional activator of several genes necessary for aerobic glycolysis.[8] More research on the mechanism(s) of the tumor suppressor function of FLCN is required.
References:
- Vocke CD, Yang Y, Pavlovich CP, et al.: High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst 97 (12): 931-5, 2005.
- Baba M, Hong SB, Sharma N, et al.: Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A 103 (42): 15552-7, 2006.
- Hasumi H, Baba M, Hong SB, et al.: Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene 415 (1-2): 60-7, 2008.
- Shaw RJ: LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 196 (1): 65-80, 2009.
- Nahorski MS, Reiman A, Lim DH, et al.: Birt Hogg-Dubé syndrome-associated FLCN mutations disrupt protein stability. Hum Mutat 32 (8): 921-9, 2011.
- Baba M, Furihata M, Hong SB, et al.: Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst 100 (2): 140-54, 2008.
- Hasumi Y, Baba M, Ajima R, et al.: Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A 106 (44): 18722-7, 2009.
- Yan M, Gingras MC, Dunlop EA, et al.: The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J Clin Invest 124 (6): 2640-50, 2014.
Clinical Manifestations
The three major features of Birt-Hogg-Dubé syndrome (BHD) include fibrofolliculomas /trichodiscomas, pulmonary cysts and spontaneous pneumothorax, and renal tumors.[1,2,3]
Cutaneous Lesions
Individuals with BHD usually present with multiple, small, skin-colored, dome-shaped papules distributed over the face, neck, and upper trunk. The characteristic dermatologic manifestation is a fibrofolliculoma or trichodiscoma (hamartoma of the hair follicle).[4] The age at diagnosis of cutaneous lesions ranges from 20 to 72 years (median age, 54 y). Only a small percentage of carriers of FLCNpathogenic variants lack cutaneous manifestations,[1,5,6] suggesting that this syndromic phenotype is highly penetrant in affected individuals. In two large BHD family studies, 73% and 84% of affected patients in whom skin lesions were biopsied were found to have fibrofolliculomas/trichodiscomas.[1,2]
Histologically, fibrofolliculomas/trichodiscomas are characterized by multiple anastomosing epithelial strands emanating from a central follicle. Mucin-rich or thick connective tissue stroma may encapsulate the epithelial component.[7] Some describe these as lesions that emanate from the sebaceous mantle of the hair follicle. The underlying molecular mechanism, which stems from FLCN loss and drives the development of fibrofolliculomas/trichodiscomas, is unclear, but one report suggests that increased WNT signaling may play a role.[7] Fibrofolliculomas and trichodiscomas are different stages of a single pathologic process.
Pulmonary Cysts and Spontaneous Pneumothorax
Computed tomography (CT) imaging results showed that lung cysts are present in 85% to 87% of patients with BHD.[1,2] These cysts are often bilateral and multifocal and are located predominantly within the lower lobes of the lung. Most BHD-related lung cysts are asymptomatic; however, individuals affected with BHD have an increased risk of developing spontaneous pneumothorax. Patients with a pathogenic variant in FLCN and a family history of spontaneous pneumothorax had a statistically significant increased risk of spontaneous pneumothorax compared with BHD patients without a family history of spontaneous pneumothorax (P = .011).[8]
In a study of 198 patients with BHD, spontaneous pneumothorax occurrences were comparable between men (20%) and women (29%). Initial pneumothoraxes occurred between the ages of 22 and 75 years.[8] However, patients had their first pneumothorax occurrences at a median age of 38 years, and these typically occurred before the fifth decade of life. Patients had a 6% chance of developing their first spontaneous pneumothoraxes by age 30 years (95% confidence interval [CI], 3%–10%). This probability increased to 75% by age 50 years (95% CI, 19%–32%).[8] A review of English PubMed literature analyzed data from 1,038 patients with BHD, and pneumothoraxes occurred in 50.9% of patients. Pneumothorax incidence seemed to differ between individuals of varying ancestries.[9]
The clinical presentation of spontaneous pneumothorax ranges from asymptomatic to dyspnea and chest pain. Clinical findings include tachypnea or decreased-to-absent breath sounds. Radiographic investigation may require a high-resolution chest CT to confirm the diagnosis because a chest x-ray may not be sensitive enough to detect a loculated pneumothorax. Up to 75% of patients with a history of spontaneous pneumothorax experience a second one. Differences in reported spontaneous pneumothorax recurrence may reflect the efficacy of different treatment modalities.
Histologic findings of pleuropulmonary lesions associated with BHD patients include thin-walled pleural and subpleural cysts and bullae, intraparenchymal air cysts, pleural blebs and changes consistent with spontaneous pneumothorax, and underlying emphysematous changes in lung tissue parenchyma adjacent to the bullae.[5]
Renal Tumors
Approximately 25% to 35% of individuals with BHD develop renal tumors,[1,4,10,11] which are multifocal in 65% of cases and often bilateral. The frequency of renal tumors among patients with BHD whose medical records were reviewed was 20%, and the frequency of renal tumors among BHD patients evaluated by CT scan was 29%. Most renal tumors associated with BHD are slow growing. Median age at diagnosis is 48 to 50 years (range, 31–71 y).[2,12,13,14] Men developed renal tumors more often than did women (27 males; 11 females). Renal tumors associated with BHD seem to occur at a younger age than do sporadic forms of renal cell cancer (RCC), in which the median age at diagnosis is 64 years.[15]Figure 1 depicts bilateral renal tumors in a patient with BHD.
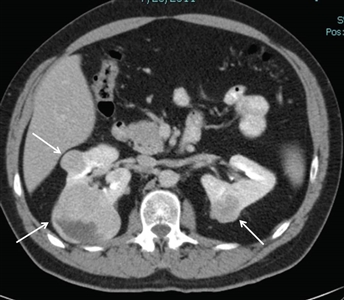
Figure 1. Birt-Hogg-Dubé syndrome–associated renal tumors are commonly multifocal and bilateral. Arrows indicate the locations of the tumors.
The most common tumors are a hybrid of oncocytoma and chromophobe histologic cell types, so-called oncocytic hybrid tumors, chromophobe RCC, and renal oncocytoma. Only renal oncocytoma is considered a benign tumor.[12] Other histologic renal tumor subtypes, including clear cell renal cell cancer (ccRCC) and papillary renal carcinoma, are uncommon in patients with BHD.[11]
Among 70 BHD patients with renal tumors and an FLCN pathogenic variant seen at the National Institutes of Health and identified through a literature review, 5 (7%) reportedly died from metastatic RCC.[1] The tumor histology in these five patients included clear cell, tubulopapillary, and/or papillary features, which are known to have a more biologically aggressive natural history. Death related to BHD-related oncocytoma and chromophobe neoplasms is exceedingly uncommon. Similar to von Hippel-Lindau disease and hereditary papillary renal carcinoma, the renal parenchyma of BHD patients commonly shows microscopic renal tumors adjacent to RCCs. The presence of microscopic oncocytosis provides histologic evidence that patients with BHD have a lifetime risk of developing renal tumors. The high frequency of FLCN somatic "second hits" (70%) in BHD-associated renal tumors supports the hypothesis that FLCN functions as a tumor suppressor gene.[16] Acquired somatic FLCNmutations have been only rarely identified in sporadic ccRCC.[17,18]
Other Manifestations
Bilateral multifocal parotid oncocytomas [19] have been reported in eight patients with BHD.[1,2,19,20,21] The bilateral, multifocal presentation of these rare tumors, in combination with recent molecular investigations, has led to the speculation that parotid oncocytomas might be part of the BHD phenotypic spectrum.
It should be noted that germline FLCNvariants were also found in patients without cutaneous findings but suspected of having BHD because of their specific renal and pulmonary manifestations.[21]
Lipomas, angiolipomas,[22] collagenomas,[4] cutaneous neurothekeomas, meningiomas,[23] multinodular goiters of thyroid,[24,25] ovarian cysts,[25] parathyroid adenomas,[22] pulmonary histiocytomas,[26] and chorioretinal lesions [25,27] have all been reported in patients with BHD. Whether these manifestations are truly associated with BHD remains to be determined.
Although initial epidemiologic observations linked BHD to an increased risk for colon polyps, subsequent epidemiologic studies did not confirm this association.[10,18,28] In a study from the Netherlands, individuals with BHD did not have increased colorectal cancer (CRC) prevalence when compared with their unaffected relatives, despite individuals with BHD participating in enhanced CRC screening.[29]
References:
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 12 (10): 558-69, 2015.
- Toro JR, Glenn G, Duray P, et al.: Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135 (10): 1195-202, 1999.
- Graham RB, Nolasco M, Peterlin B, et al.: Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 172 (1): 39-44, 2005.
- Painter JN, Tapanainen H, Somer M, et al.: A 4-bp deletion in the Birt-Hogg-Dubé gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet 76 (3): 522-7, 2005.
- Vernooij M, Claessens T, Luijten M, et al.: Birt-Hogg-Dubé syndrome and the skin. Fam Cancer 12 (3): 381-5, 2013.
- Toro JR, Pautler SE, Stewart L, et al.: Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 175 (10): 1044-53, 2007.
- Matsumoto K, Lim D, Pharoah PD, et al.: A systematic review assessing the existence of pneumothorax-only variants of FLCN. Implications for lifelong surveillance of renal tumours. Eur J Hum Genet 29 (11): 1595-1600, 2021.
- Zbar B, Alvord WG, Glenn G, et al.: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 11 (4): 393-400, 2002.
- Pavlovich CP, Walther MM, Eyler RA, et al.: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 26 (12): 1542-52, 2002.
- Pavlovich CP, Grubb RL, Hurley K, et al.: Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol 173 (5): 1482-6, 2005.
- Shuch B, Vourganti S, Ricketts CJ, et al.: Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 32 (5): 431-7, 2014.
- Sattler EC, Reithmair M, Steinlein OK: Kidney cancer characteristics and genotype-phenotype-correlations in Birt-Hogg-Dubé syndrome. PLoS One 13 (12): e0209504, 2018.
- Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2017. Bethesda, Md: National Cancer Institute, 2020. Available online. Last accessed August 9, 2024.
- Vocke CD, Yang Y, Pavlovich CP, et al.: High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst 97 (12): 931-5, 2005.
- da Silva NF, Gentle D, Hesson LB, et al.: Analysis of the Birt-Hogg-Dubé (BHD) tumour suppressor gene in sporadic renal cell carcinoma and colorectal cancer. J Med Genet 40 (11): 820-4, 2003.
- Khoo SK, Kahnoski K, Sugimura J, et al.: Inactivation of BHD in sporadic renal tumors. Cancer Res 63 (15): 4583-7, 2003.
- Liu V, Kwan T, Page EH: Parotid oncocytoma in the Birt-Hogg-Dubé syndrome. J Am Acad Dermatol 43 (6): 1120-2, 2000.
- Vinit J, Friedel J, Bielefeld P, et al.: [Birt-Hogg-Dubé syndrome and multiple recurrent tumors]. Rev Med Interne 32 (3): e40-2, 2011.
- Maffé A, Toschi B, Circo G, et al.: Constitutional FLCN mutations in patients with suspected Birt-Hogg-Dubé syndrome ascertained for non-cutaneous manifestations. Clin Genet 79 (4): 345-54, 2011.
- Chung JY, Ramos-Caro FA, Beers B, et al.: Multiple lipomas, angiolipomas, and parathyroid adenomas in a patient with Birt-Hogg-Dube syndrome. Int J Dermatol 35 (5): 365-7, 1996.
- Vincent A, Farley M, Chan E, et al.: Birt-Hogg-Dubé syndrome: two patients with neural tissue tumors. J Am Acad Dermatol 49 (4): 717-9, 2003.
- Drummond C, Grigoris I, Dutta B: Birt-Hogg-Dubé syndrome and multinodular goitre. Australas J Dermatol 43 (4): 301-4, 2002.
- Welsch MJ, Krunic A, Medenica MM: Birt-Hogg-Dubé Syndrome. Int J Dermatol 44 (8): 668-73, 2005.
- Tomassetti S, Carloni A, Chilosi M, et al.: Pulmonary features of Birt-Hogg-Dubé syndrome: cystic lesions and pulmonary histiocytoma. Respir Med 105 (5): 768-74, 2011.
- Walter P, Kirchhof B, Korge B, et al.: Flecked chorioretinopathy associated with Birt-Hogg-Dubé syndrome. Graefes Arch Clin Exp Ophthalmol 235 (6): 359-61, 1997.
- Nahorski MS, Lim DH, Martin L, et al.: Investigation of the Birt-Hogg-Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J Med Genet 47 (6): 385-90, 2010.
- van de Beek I, Glykofridis IE, Wolthuis RMF, et al.: No evidence for increased prevalence of colorectal carcinoma in 399 Dutch patients with Birt-Hogg-Dubé syndrome. Br J Cancer 122 (4): 590-594, 2020.
Risk Assessment for Birt-Hogg-Dubé Syndrome (BHD)
Genetic Testing
FLCN is the only gene known to be associated with BHD. It is located on chromosome 17p11.2.[1] Molecular testing is available for clinical applications such as diagnostic testing and prenatal diagnosis. Of families with BHD, 53% (27 of 51) were found to have an insertion or deletion in the polycytosine tract in exon 11 (a variant hot spot).[2] Bidirectional DNA sequencing of all FLCN-coding exons (exon 4–14) resulted in a pathogenic variant detection rate of 84%.[2,3] This rate has been further improved by the development of real time-quantitative polymerase chain reaction and multiplex ligation-dependent probe amplification assays to detect intragenic deletions and duplications;[4] these assays are available on a clinical basis.
Genetic testing performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory is indicated for all individuals known to have or suspected of having BHD, including individuals with the following:
- Five or more facial or truncal papules with at least one histologically confirmed fibrofolliculoma [5] with or without family history of BHD.
- A family history of BHD with a single fibrofolliculoma or a single renal tumor or history of spontaneous pneumothorax.
- Multiple and bilateral chromophobe, and/or oncocytic hybrid renal tumors.
- A single chromophobe, or oncocytic hybrid tumor and a family history of renal cancer with any of above renal cell tumor types.
- A family history of autosomal dominant primary spontaneous pneumothorax without a history of lung cyst.
Genetic Counseling
BHD syndrome is inherited in an autosomal dominant manner. If a parent of a proband is clinically affected or has a FLCN pathogenic variant, the siblings of the proband have a 50% chance of inheriting the variant. The degree of clinical severity is not predictable. Prenatal diagnosis of BHD is possible in pregnancies that have a 50% chance of inheriting a FLCN pathogenic variant if the disease-causing variant has been identified in an affected family member.
The Cancer Genetics Risk Assessment and Counseling summary can provide more information about the following topics:
Clinical Diagnosis
The three major features of BHD include cutaneous lesions, lung manifestations (lung cysts and spontaneous pneumothoraxes), and renal tumors.[2,3] For more information about these BHD manifestations, see the Clinical Manifestations section.
The dermatologic diagnosis of BHD is made in individuals who have five or more facial or truncal papules with at least one histologically confirmed fibrofolliculoma.[5] An adequate biopsy (typically a punch biopsy) is required to make a diagnosis of fibrofolliculoma. An expert panel has developed the following diagnostic criteria for BHD (patients must fulfill one major or two minor criteria for diagnosis):[6]
- Major criteria:
- At least five fibrofolliculomas/trichodiscomas, at least one histologically confirmed, of adult onset.
- Germline FLCN pathogenic variant.
- Minor criteria:
- Multiple lung cysts: bilateral, basally located lung cysts with no other apparent cause, with or without spontaneous primary pneumothorax.
- Renal cancer: early-onset (age <50 y) or multifocal or bilateral renal cancer, or renal cancer of mixed chromophobe and oncocytic histology.
- A first-degree relative with BHD.
Differential diagnosis
It is important to distinguish between BHD-associated RCC and sporadic RCC because this may have implications for management. Genetic testing for a pathogenic variant in FLCN, a family history of BHD, or the presence of extrarenal manifestations associated with BHD are helpful in establishing a diagnosis of this condition. Because a variety of histological variants of kidney cancer can be seen in association with BHD, it is often necessary to make a histological diagnosis to help differentiate between the benign tumors (oncocytomas) and those with a malignant potential (chromophobe, clear cell, and papillary RCC).[7]
The differential diagnosis of pulmonary cysts includes lymphangioleiomyomatosis (LAM); distinguishing this from BHD can be clinically challenging. One study proposed a set of findings that permit differentiation between BHD and LAM.[8] These include bibasilar, peripheral, and subpleural distribution for BHD versus diffuse distribution for LAM; elliptical or lentiform shape for BHD-related cysts versus round shape for LAM; and HMB-45 negativity on immunohistochemical staining for BHD versus HMB-45 positivity for LAM. This approach has not been validated; further study is warranted.
References:
- Schmidt LS, Warren MB, Nickerson ML, et al.: Birt-Hogg-Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet 69 (4): 876-82, 2001.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Benhammou JN, Vocke CD, Santani A, et al.: Identification of intragenic deletions and duplication in the FLCN gene in Birt-Hogg-Dubé syndrome. Genes Chromosomes Cancer 50 (6): 466-77, 2011.
- Toro JR, Glenn G, Duray P, et al.: Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135 (10): 1195-202, 1999.
- Menko FH, van Steensel MA, Giraud S, et al.: Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol 10 (12): 1199-206, 2009.
- Pavlovich CP, Grubb RL, Hurley K, et al.: Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol 173 (5): 1482-6, 2005.
- Gupta N, Seyama K, McCormack FX: Pulmonary manifestations of Birt-Hogg-Dubé syndrome. Fam Cancer 12 (3): 387-96, 2013.
Surveillance for Birt-Hogg-Dubé Syndrome (BHD)
BHD patients display two main clinical presentations. Most commonly, individuals present with a documented family history of BHD. Other presentations include individuals without a BHD family history or one that is unknown. In the former clinical scenario, if the patient's biological relative has a genetic diagnosis with an identified FLCN pathogenic variant, the patient may choose to begin evaluation with genetic counseling and pathogenic variant testing.
Clinical surveillance for individuals at risk of BHD includes dermatologic, radiological, and histological examinations to identify characteristic cutaneous lesions, renal tumors, and lung cysts, with or without a history of spontaneous pneumothorax. Not all features are present in each at-risk individual, and some BHD family members may have no discernible phenotypic findings (i.e., they are clinically unaffected carriers of deleterious FLCN variants). This clinical scenario is being encountered with increasing frequency as the number of syndrome-associated genes for which pathogenic variant testing can be offered clinically expands. In most disorders, the natural history of genetically abnormal/clinically normal individuals has not yet been well characterized. These major features of BHD are described in the Clinical diagnosis section.
Decisions regarding the use of lifelong surveillance for hereditary RCC syndromes must consider both risks and benefits. Approximately 15% to 29% of individuals with BHD have renal tumors,[1,2] which are commonly bilateral and multifocal and include a number of specific histologies within an individual or family.[3] For at-risk individuals who will undergo periodic imaging for many years even when no tumor is present, a surveillance schedule that minimizes the lifetime dose of radiation is advised.
Contrast-enhanced computed tomography (CT) or magnetic resonance imaging (MRI) are both useful modalities for the detection of BHD-associated renal tumors.[3] Ultrasonography alone may not be sufficient for detecting renal tumors because some tumors are isoechoic with the renal parenchyma,[4] but they may help identify renal cysts. A series in the Netherlands failed to detect 9 of 18 renal tumors using ultrasonography alone. Thus, while ultrasonography may reliably detect larger lesions, it is not a reliable modality for detecting smaller lesions and is therefore not used routinely as a lone screening tool.[5] If a renal tumor is detected, the patient is referred to a urological oncology surgeon for management, which may include continued monitoring or surgery, depending mainly on tumor size.[3] If no renal tumor is detected on initial imaging, experts recommend lifelong surveillance at least once every 36 months because of the risk of developing RCC.[4] Because MRI spares the patient from exposure to radiation, it is reasonable to assume that it may be the preferred mode of imaging over CT for lifelong surveillance.
Level of evidence: 5
References:
- Zbar B, Alvord WG, Glenn G, et al.: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 11 (4): 393-400, 2002.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Pavlovich CP, Grubb RL, Hurley K, et al.: Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol 173 (5): 1482-6, 2005.
- Stamatakis L, Metwalli AR, Middelton LA, et al.: Diagnosis and management of BHD-associated kidney cancer. Fam Cancer 12 (3): 397-402, 2013.
- Johannesma PC, van de Beek I, van der Wel TJWT, et al.: Renal imaging in 199 Dutch patients with Birt-Hogg-Dubé syndrome: Screening compliance and outcome. PLoS One 14 (3): e0212952, 2019.
Management of Disease Manifestations for Birt-Hogg-Dubé Syndrome (BHD)
Management of Skin Manifestations
Cryotherapy, electrodessication, surgery, and laser therapy have been used with good cosmetic results, but relapse usually occurs because the cutaneous lesions are a manifestation of an inherited skin condition.[1,2,3] Therefore, patients may require continuous cosmetic care. Some patients with BHD are emotionally affected by their dermatologic condition, regardless of the number or extent of cutaneous lesions. Therefore, the psychological state of patients with BHD warrants consideration, with skin care recommendations appropriately tailored to individual needs.
Level of evidence: 5
Management of Renal Manifestations
Partial nephrectomy is the treatment of choice in the management of BHD-related kidney neoplasms, to preserve optimal long-term kidney function in patients at risk of multiple primary renal tumors. However, this renal-sparing surgery depends on the size and location of the tumors found during surgery. It is important to incorporate knowledge of the high cumulative risk of multifocal and bilateral kidney tumors in this syndrome, as surgical management is planned. In general, renal tumors smaller than 3 cm in diameter may be monitored radiologically under the close supervision of the urological oncology surgeon; immediate surgery may not be required.[4] These are general recommendations, and each case should be evaluated carefully and managed individually. Total nephrectomy may be necessary in some cases.
Surveillance of at-risk individuals and relatives includes abdominal/pelvic MRI or CT scans and evaluation of renal tumors by urological surgeons and radiologists experienced in the management of these complicated patients. Use of genetic testing for early identification of at-risk family members improves diagnostic certainty and eliminates costly and stressful screening procedures in at-risk relatives who have not inherited their family's disease-causing variant.
Level of evidence: 4
Management of Spontaneous Pneumothorax
The management of spontaneous pneumothorax in patients with BHD is similar to that employed in the general population.[5]
The clinical presentation of spontaneous pneumothorax in patients with BHD is variable. Therapy is dictated by the underlying lung condition and general health of the patient. One study reported that of 101 patients with spontaneous pneumothoraces, 78 required medical intervention, and 23 were managed by observation alone.[5] Thirty-five percent of patients with pneumothoraces were treated with tube thoracostomy (chest tube) only; 14% were treated by open thoracotomy and a second treatment, including mechanical or chemical pleurodesis and lung resection; and approximately 13% were treated with combined tube thoracostomy, thoracotomy, and a third treatment, including mechanical or chemical pleurodesis or lung resection. Patients with BHD—especially those with multiple lung cysts—should be advised to avoid or be cautious with scuba diving, air travel, and mechanical ventilation because each exposure increases the risk of spontaneous pneumothorax.[5]
Level of evidence: 4
References:
- Jacob CI, Dover JS: Birt-Hogg-Dube syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol 137 (1): 98-9, 2001.
- Gambichler T, Wolter M, Altmeyer P, et al.: Treatment of Birt-Hogg-Dubé syndrome with erbium:YAG laser. J Am Acad Dermatol 43 (5 Pt 1): 856-8, 2000.
- Kahle B, Hellwig S, Schulz T: [Multiple mantleomas in Birt-Hogg-Dubé syndrome: successful therapy with CO2 laser] Hautarzt 52 (1): 43-6, 2001.
- Pavlovich CP, Grubb RL, Hurley K, et al.: Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol 173 (5): 1482-6, 2005.
- Toro JR, Pautler SE, Stewart L, et al.: Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 175 (10): 1044-53, 2007.
Prognosis
The major cause of morbidity and mortality in Birt-Hogg-Dubé syndrome (BHD) is related to renal lesions. Because of the rarity of BHD, it is difficult to generate robust overall survival data on populations of patients with the syndrome; however, when patients are managed with an appropriate surveillance and intervention strategy, their life expectancy should not be significantly different from that of matched individuals in the general population.
While a majority of patients have excellent outcomes when tumors are detected early and removed surgically, there is a risk of metastasis with larger tumors; the optimal management for metastatic disease is unclear.[1]
References:
- Johannesma PC, van de Beek I, van der Wel TJWT, et al.: Renal imaging in 199 Dutch patients with Birt-Hogg-Dubé syndrome: Screening compliance and outcome. PLoS One 14 (3): e0212952, 2019.
Future Directions
Since FLCN, the gene responsible for Birt-Hogg-Dubé syndrome, was identified in 2001, a number of studies have elucidated its function and possible genotype -phenotype correlations. Although surveillance followed by surgical resection remains the mainstay of disease management, improvements in early detection and in molecularly targeted early intervention may alter the course of this disease in the kidney and decrease the incidence of overt and/or lethal renal manifestations. A better understanding of the biochemical function of the FLCN protein should provide insights into target identification and validation of medical therapy for localized, locally advanced, and metastatic disease.
Latest Updates to This Summary (12 / 16 / 2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
The summary was extensively revised.
This summary is written and maintained by the PDQ Cancer Genetics Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the genetics of Birt-Hogg-Dubé syndrome. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Cancer Genetics Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Birt-Hogg-Dubé Syndrome are:
- Alexandra Perez Lebensohn, MS, CGC (National Cancer Institute)
- Brian Matthew Shuch, MD (UCLA Health)
- Ramaprasad Srinivasan, MD, PhD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Cancer Genetics Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Cancer Genetics Editorial Board. PDQ Birt-Hogg-Dubé Syndrome. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/publications/pdq/information-summaries/genetics/bhd-syndrome-hp-pdq. Accessed <MM/DD/YYYY>. [PMID: 33724752]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
The information in these summaries should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-12-16

